NeuroCult™ Chemical Dissociation Kit (Mouse)
Kit for chemical dissociation of mouse neurospheres
概要
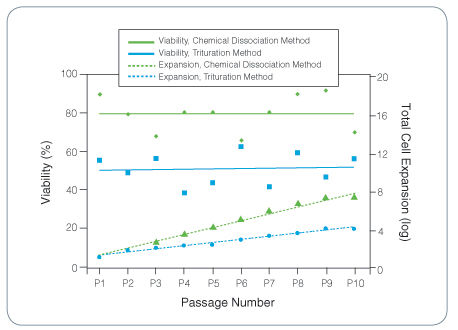
The NeuroCult™ Chemical Dissociation Kit is recommended for the non-mechanical and non-enzymatic dissociation of neurospheres derived from embryonic or adult mouse central nervous system tissue. A significantly higher viability and total cell number is observed after expansion, in comparison to neurospheres dissociated by trituration, and the functional properties of the cells are maintained.
- NeuroCult™ Chemical Dissociation Solution A, 55 mL
- NeuroCult™ Chemical Dissociation Solution B, 15 mL
- NeuroCult™ Chemical Dissociation Solution C, 15 mL
Neural Stem and Progenitor Cells
Neuroscience, Stem Cell Biology
数据及文献
Publications (35)
Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology 2017 JUN
Cathepsin B Improves ß-Amyloidosis and Learning and Memory in Models of Alzheimer's Disease.
Embury CM et al.
Abstract
Amyloid-ß (Aß) precursor protein (APP) metabolism engages neuronal endolysosomal pathways for Aß processing and secretion. In Alzheimer's disease (AD), dysregulation of APP leads to excess Aß and neuronal dysfunction; suggesting that neuronal APP/Aß trafficking can be targeted for therapeutic gain. Cathepsin B (CatB) is a lysosomal cysteine protease that can lower Aß levels. However, whether CatB-modulation of Aß improves learning and memory function deficits in AD is not known. To this end, progenitor neurons were infected with recombinant adenovirus expressing CatB and recovered cell lysates subjected to proteomic analyses. The results demonstrated Lamp1 deregulation and linkages between CatB and the neuronal phagosome network. Hippocampal injections of adeno-associated virus expressing CatB reduced Aß levels, increased Lamp1 and improved learning and memory. The findings were associated with the emergence of c-fos + cells. The results support the idea that CatB can speed Aß metabolism through lysosomal pathways and as such reduce AD-associated memory deficits.
Aging cell 2017 FEB
DNA polymerase β decrement triggers death of olfactory bulb cells and impairs olfaction in a mouse model of Alzheimer's disease.
Misiak M et al.
Abstract
Alzheimer's disease (AD) involves the progressive degeneration of neurons critical for learning and memory. In addition, patients with AD typically exhibit impaired olfaction associated with neuronal degeneration in the olfactory bulb (OB). Because DNA base excision repair (BER) is reduced in brain cells during normal aging and AD, we determined whether inefficient BER due to reduced DNA polymerase-β (Polβ) levels renders OB neurons vulnerable to degeneration in the 3xTgAD mouse model of AD. We interrogated OB histopathology and olfactory function in wild-type and 3xTgAD mice with normal or reduced Polβ levels. Compared to wild-type control mice, Polβ heterozygous (Polβ+/- ), and 3xTgAD mice, 3xTgAD/Polβ+/- mice exhibited impaired performance in a buried food test of olfaction. Polβ deficiency did not affect the proliferation of OB neural progenitor cells in the subventricular zone. However, numbers of newly generated neurons were reduced by approximately 25% in Polβ+/- and 3xTgAD mice, and by over 60% in the 3xTgAD/Polβ+/- mice compared to wild-type control mice. Analyses of DNA damage and apoptosis revealed significantly greater degeneration of OB neurons in 3xTgAD/Polβ+/- mice compared to 3xTgAD mice. Levels of amyloid β-peptide (Aβ) accumulation in the OB were similar in 3xTgAD and 3xTgAD/Polβ+/- mice, and cultured Polβ-deficient neurons exhibited increased vulnerability to Aβ-induced death. Olfactory deficit is an early sign in human AD, but the mechanism is not yet understood. Our findings in a new AD mouse model demonstrate that diminution of BER can endanger OB neurons, and suggest a mechanism underlying early olfactory impairment in AD.
EMBO molecular medicine 2016 MAR
Therapeutic potential of targeting microRNA-10b in established intracranial glioblastoma: first steps toward the clinic.
Teplyuk NM et al.
Abstract
MicroRNA-10b (miR-10b) is a unique oncogenic miRNA that is highly expressed in all GBM subtypes, while absent in normal neuroglial cells of the brain. miR-10b inhibition strongly impairs proliferation and survival of cultured glioma cells, including glioma-initiating stem-like cells (GSC). Although several miR-10b targets have been identified previously, the common mechanism conferring the miR-10b-sustained viability of GSC is unknown. Here, we demonstrate that in heterogeneous GSC, miR-10b regulates cell cycle and alternative splicing, often through the non-canonical targeting via 5'UTRs of its target genes, including MBNL1-3, SART3, and RSRC1. We have further assessed the inhibition of miR-10b in intracranial human GSC-derived xenograft and murine GL261 allograft models in athymic and immunocompetent mice. Three delivery routes for the miR-10b antisense oligonucleotide inhibitors (ASO), direct intratumoral injections, continuous osmotic delivery, and systemic intravenous injections, have been explored. In all cases, the treatment with miR-10b ASO led to targets' derepression, and attenuated growth and progression of established intracranial GBM. No significant systemic toxicity was observed upon ASO administration by local or systemic routes. Our results indicate that miR-10b is a promising candidate for the development of targeted therapies against all GBM subtypes.
Stem cell reports 2016 MAR
EVA1A/TMEM166 Regulates Embryonic Neurogenesis by Autophagy.
Li M et al.
Abstract
Self-renewal and differentiation of neural stem cells is essential for embryonic neurogenesis, which is associated with cell autophagy. However, the mechanism by which autophagy regulates neurogenesis remains undefined. Here, we show that Eva1a/Tmem166, an autophagy-related gene, regulates neural stem cell self-renewal and differentiation. Eva1a depletion impaired the generation of newborn neurons, both in vivo and in vitro. Conversely, overexpression of EVA1A enhanced newborn neuron generation and maturation. Moreover, Eva1a depletion activated the PIK3CA-AKT axis, leading to the activation of the mammalian target of rapamycin and the subsequent inhibition of autophagy. Furthermore, addition of methylpyruvate to the culture during neural stem cell differentiation rescued the defective embryonic neurogenesis induced by Eva1a depletion, suggesting that energy availability is a significant factor in embryonic neurogenesis. Collectively, these data demonstrated that EVA1A regulates embryonic neurogenesis by modulating autophagy. Our results have potential implications for understanding the pathogenesis of neurodevelopmental disorders caused by autophagy dysregulation.
Journal of virology 2016 JAN
Ecotropic Murine Leukemia Virus Infection of Glial Progenitors Interferes with Oligodendrocyte Differentiation: Implications for Neurovirulence.
Li Y et al.
Abstract
UNLABELLED Certain murine leukemia viruses (MLVs) are capable of inducing fatal progressive spongiform motor neuron disease in mice that is largely mediated by viral Env glycoprotein expression within central nervous system (CNS) glia. While the etiologic mechanisms and the glial subtypes involved remain unresolved, infection of NG2 glia was recently observed to correlate spatially and temporally with altered neuronal physiology and spongiogenesis. Since one role of NG2 cells is to serve as oligodendrocyte (OL) progenitor cells (OPCs), we examined here whether their infection by neurovirulent (FrCasE) or nonneurovirulent (Fr57E) ecotropic MLVs influenced their viability and/or differentiation. Here, we demonstrate that OPCs, but not OLs, are major CNS targets of both FrCasE and Fr57E. We also show that MLV infection of neural progenitor cells (NPCs) in culture did not affect survival, proliferation, or OPC progenitor marker expression but suppressed certain glial differentiation markers. Assessment of glial differentiation in vivo using transplanted transgenic NPCs showed that, while MLVs did not affect cellular engraftment or survival, they did inhibit OL differentiation, irrespective of MLV neurovirulence. In addition, in chimeric brains, where FrCasE-infected NPC transplants caused neurodegeneration, the transplanted NPCs proliferated. These results suggest that MLV infection is not directly cytotoxic to OPCs but rather acts to interfere with OL differentiation. Since both FrCasE and Fr57E viruses restrict OL differentiation but only FrCasE induces overt neurodegeneration, restriction of OL maturation alone cannot account for neuropathogenesis. Instead neurodegeneration may involve a two-hit scenario where interference with OPC differentiation combined with glial Env-induced neuronal hyperexcitability precipitates disease. IMPORTANCE A variety of human and animal retroviruses are capable of causing central nervous system (CNS) neurodegeneration manifested as motor and cognitive deficits. These retroviruses infect a variety of CNS cell types; however, the specific role each cell type plays in neuropathogenesis remains to be established. The NG2 glia, whose CNS functions are only now emerging, are a newly appreciated viral target in murine leukemia virus (MLV)-induced neurodegeneration. Since one role of NG2 glia is that of oligodendrocyte progenitor cells (OPCs), we investigated here whether their infection by the neurovirulent MLV FrCasE contributed to neurodegeneration by affecting OPC viability and/or development. Our results show that both neurovirulent and nonneurovirulent MLVs interfere with oligodendrocyte differentiation. Thus, NG2 glial infection could contribute to neurodegeneration by preventing myelin formation and/or repair and by suspending OPCs in a state of persistent susceptibility to excitotoxic insult mediated by neurovirulent virus effects on other glial subtypes.
Frontiers in cellular neuroscience 2016 DEC
Heterocellular Contacts with Mouse Brain Endothelial Cells Via Laminin and α6β1 Integrin Sustain Subventricular Zone (SVZ) Stem/Progenitor Cells Properties.
Rosa AI et al.
Abstract
Neurogenesis in the subventricular zone (SVZ) is regulated by diffusible factors and cell-cell contacts. In vivo, SVZ stem cells are associated with the abluminal surface of blood vessels and such interactions are thought to regulate their neurogenic capacity. SVZ neural stem cells (NSCs) have been described to contact endothelial-derived laminin via α6β1 integrin. To elucidate whether heterocellular contacts with brain endothelial cells (BEC) regulate SVZ cells neurogenic capacities, cocultures of SVZ neurospheres and primary BEC, both obtained from C57BL/6 mice, were performed. The involvement of laminin-integrin interactions in SVZ homeostasis was tested in three ways. Firstly, SVZ cells were analyzed following incubation of BEC with the protein synthesis inhibitor cycloheximide (CHX) prior to coculture, a treatment expected to decrease membrane proteins. Secondly, SVZ cells were cocultured with BEC in the presence of an anti-α6 integrin neutralizing antibody. Thirdly, BEC were cultured with β1-/- SVZ cells. We showed that contact with BEC supports, at least in part, proliferation and stemness of SVZ cells, as evaluated by the number of BrdU positive (+) and Sox2+ cells in contact with BEC. These effects are dependent on BEC-derived laminin binding to α6β1 integrin and are decreased in cocultures incubated with anti-α6 integrin neutralizing antibody and in cocultures with SVZ β1-/- cells. Moreover, BEC-derived laminin sustains stemness in SVZ cell cultures via activation of the Notch and mTOR signaling pathways. Our results show that BEC/SVZ interactions involving α6β1 integrin binding to laminin, contribute to SVZ cell proliferation and stemness.
View All Publications