
FK-866
Nicotinamide phosphoribosyltransferase inhibitor


概要
FK-866 is a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme that regulates NAD+ biosynthesis from the natural precursor nicotinamide (Cameron et al.; Hasmann & Schemainda). In hepatocarcinoma cells, FK-866 activates AMP-activated protein kinase (AMPK) and downregulates mammalian target of rapamycin (mTOR) signaling (Schuster et al.).
METABOLISM
· FK-866 can be used to deplete NAD+, a central metabolism cofactor (Cameron et al.; Hasmann & Schemainda; Jadeja et al.).
CANCER RESEARCH
· Depletes NAD+ and induces delayed cell death by apoptosis in HepG2 human liver carcinoma cells (IC₅₀ = ~1 nM; Hasmann & Schemainda).
· Triggers dose-dependent cytotoxicity in multiple myeloma cells (Cea et al.).
· Induces autophagic death in neuroblastoma SH-SY5Y cells (IC₅₀ = 0.93 nM; Billington et al.).
METABOLISM
· FK-866 can be used to deplete NAD+, a central metabolism cofactor (Cameron et al.; Hasmann & Schemainda; Jadeja et al.).
CANCER RESEARCH
· Depletes NAD+ and induces delayed cell death by apoptosis in HepG2 human liver carcinoma cells (IC₅₀ = ~1 nM; Hasmann & Schemainda).
· Triggers dose-dependent cytotoxicity in multiple myeloma cells (Cea et al.).
· Induces autophagic death in neuroblastoma SH-SY5Y cells (IC₅₀ = 0.93 nM; Billington et al.).
Alternative Names
K 22.175
Area of Interest
Autophagy, Cancer Research, Metabolism
CAS Number
658084-64-1
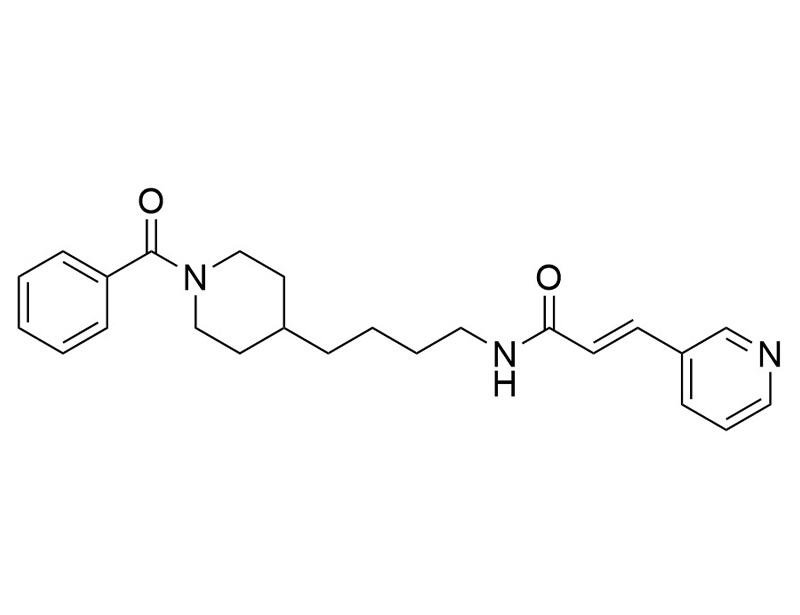
Chemical Formula
C₂₄H₂₉N₃O₂
Molecular Weight
391.5 g/mol
Purity
≥ 98%
Pathway
mTOR
Target
AMPK
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | FK-866 | 100-0263, 100-0264 | All | English |
| Safety Data Sheet | FK-866 | 100-0263, 100-0264 | All | English |
数据及文献
Publications (6)
Nature immunology 2019
Inflammatory macrophage dependence on NAD+ salvage is a consequence of reactive oxygen species-mediated DNA damage.
Abstract
Abstract
The adoption of Warburg metabolism is critical for the activation of macrophages in response to lipopolysaccharide. Macrophages stimulated with lipopolysaccharide increase their expression of nicotinamide phosphoribosyltransferase (NAMPT), a key enzyme in NAD+ salvage, and loss of NAMPT activity alters their inflammatory potential. However, the events that lead to the cells' becoming dependent on NAD+ salvage remain poorly defined. We found that depletion of NAD+ and increased expression of NAMPT occurred rapidly after inflammatory activation and coincided with DNA damage caused by reactive oxygen species (ROS). ROS produced by complex III of the mitochondrial electron-transport chain were required for macrophage activation. DNA damage was associated with activation of poly(ADP-ribose) polymerase, which led to consumption of NAD+. In this setting, increased NAMPT expression allowed the maintenance of NAD+ pools sufficient for glyceraldehyde-3-phosphate dehydrogenase activity and Warburg metabolism. Our findings provide an integrated explanation for the dependence of inflammatory macrophages on the NAD+ salvage pathway.
Aging 2018 jun
Loss of NAMPT in aging retinal pigment epithelium reduces NAD+ availability and promotes cellular senescence.
Abstract
Abstract
Retinal pigment epithelium (RPE) performs numerous functions critical to retinal health and visual function. RPE senescence is a hallmark of aging and degenerative retinal disease development. Here, we evaluated the temporal expression of key nicotinamide adenine dinucleotide (NAD+)-biosynthetic genes and associated levels of NAD+, a principal regulator of energy metabolism and cellular fate, in mouse RPE. NAD+ levels declined with age and correlated directly with decreased nicotinamide phosphoribosyltransferase (NAMPT) expression, increased expression of senescence markers (p16INK4a, p21Waf/Cip1, ApoJ, CTGF and $\beta$-galactosidase) and significant reductions in SIRT1 expression and activity. We simulated in vitro the age-dependent decline in NAD+ and the related increase in RPE senescence in human (ARPE-19) and mouse primary RPE using the NAMPT inhibitor FK866 and demonstrated the positive impact of NAD+-enhancing therapies on RPE cell viability. This, we confirmed in vivo in the RPE of mice injected sub-retinally with FK866 in the presence or absence of nicotinamide mononucleotide. Our data confirm the importance of NAD+ to RPE cell biology normally and in aging and demonstrate the potential utility of therapies targeting NAMPT and NAD+ biosynthesis to prevent or alleviate consequences of RPE senescence in aging and/or degenerative retinal diseases in which RPE dysfunction is a crucial element.
Biochemical and biophysical research communications 2015 mar
FK866-induced NAMPT inhibition activates AMPK and downregulates mTOR signaling in hepatocarcinoma cells.
Abstract
Abstract
BACKGROUND Nicotinamide phosphoribosyltransferase (NAMPT) is the key enzyme of the NAD salvage pathway starting from nicotinamide. Cancer cells have an increased demand for NAD due to their high proliferation and DNA repair rate. Consequently, NAMPT is considered as a putative target for anti-cancer therapies. There is evidence that AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) become dysregulated during the development of hepatocellular carcinoma (HCC). Here, we investigated the effects of NAMPT inhibition by its specific inhibitor FK866 on the viability of hepatocarcinoma cells and analyzed the effects of FK866 on the nutrient sensor AMPK and mTOR complex1 (mTORC1) signaling. RESULTS FK866 markedly decreased NAMPT activity and NAD content in hepatocarcinoma cells (Huh7 cells, Hep3B cells) and led to delayed ATP reduction which was associated with increased cell death. These effects could be abrogated by administration of nicotinamide mononucleotide (NMN), the enzyme product of NAMPT. Our results demonstrated a dysregulation of the AMPK/mTOR pathway in hepatocarcinoma cells compared to non-cancerous hepatocytes with a higher expression of mTOR and a lower AMPK$\alpha$ activation in hepatocarcinoma cells. We found that NAMPT inhibition by FK866 significantly activated AMPK$\alpha$ and inhibited the activation of mTOR and its downstream targets p70S6 kinase and 4E-BP1 in hepatocarcinoma cells. Non-cancerous hepatocytes were less sensitive to FK866 and did not show changes in AMPK/mTOR signaling after FK866 treatment. CONCLUSION Taken together, these findings reveal an important role of the NAMPT-mediated NAD salvage pathway in the energy homeostasis of hepatocarcinoma cells and suggest NAMPT inhibition as a potential treatment option for HCC.
Blood 2012 oct
Targeting NAD+ salvage pathway induces autophagy in multiple myeloma cells via mTORC1 and extracellular signal-regulated kinase (ERK1/2) inhibition.
Abstract
Abstract
Malignant cells have a higher nicotinamide adenine dinucleotide (NAD(+)) turnover rate than normal cells, making this biosynthetic pathway an attractive target for cancer treatment. Here we investigated the biologic role of a rate-limiting enzyme involved in NAD(+) synthesis, Nampt, in multiple myeloma (MM). Nampt-specific chemical inhibitor FK866 triggered cytotoxicity in MM cell lines and patient MM cells, but not normal donor as well as MM patients PBMCs. Importantly, FK866 in a dose-dependent fashion triggered cytotoxicity in MM cells resistant to conventional and novel anti-MM therapies and overcomes the protective effects of cytokines (IL-6, IGF-1) and bone marrow stromal cells. Nampt knockdown by RNAi confirmed its pivotal role in maintenance of both MM cell viability and intracellular NAD(+) stores. Interestingly, cytotoxicity of FK866 triggered autophagy, but not apoptosis. A transcriptional-dependent (TFEB) and independent (PI3K/mTORC1) activation of autophagy mediated FK866 MM cytotoxicity. Finally, FK866 demonstrated significant anti-MM activity in a xenograft-murine MM model, associated with down-regulation of ERK1/2 phosphorylation and proteolytic cleavage of LC3 in tumor cells. Our data therefore define a key role of Nampt in MM biology, providing the basis for a novel targeted therapeutic approach.
Autophagy 2008 apr
NAD depletion by FK866 induces autophagy.
Abstract
Abstract
NAD is a multifunctional molecule involved in both metabolic processes and signaling pathways. Such signalling pathways consume NAD which is replenished via one of several biosynthesis pathways. We show that influx of NAD across the plasma membrane may be able to contribute to the homeostasis of intracellular NAD levels. Indeed, extracellular application of NAD was able to replete NAD levels that had been lowered pharmacologically using the novel drug FK866 and was also able to rescue cells from FK866-induced cell death. A marked lag between the drop in NAD levels and cell death prompted us to investigate the mechanism of cell death. We were unable to find evidence of apoptosis as assessed by immunoblotting for the Caspase 3 activation fragment and immunostaining for cytochrome C and AIF translocation. We, therefore, investigated whether autophagy was initiated by FK866. Indeed, we were able to observe the formation of LC3-positive vesicles that had fused with lysosomes in FK866-treated but not control cells. Furthermore, this autophagic phenotype could be reverted by the addition of NAD to the extracellular medium.
Cancer research 2003 nov
FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis.
Abstract
Abstract
Deregulation of apoptosis, the physiological form of cell death, is closely associated with immunological diseases and cancer. Apoptosis is activated either by death receptor-driven or mitochondrial pathways, both of which may provide potential targets for novel anticancer drugs. Although several ligands stimulating death receptors have been described, the actual molecular events triggering the mitochondrial pathway are largely unknown. Here, we show initiation of apoptosis by gradual depletion of the intracellular coenzyme NAD+. We identified the first low molecular weight compound, designated FK866, which induces apoptosis by highly specific, noncompetitive inhibition of nicotinamide phosphoribosyltransferase (NAPRT), a key enzyme in the regulation of NAD+ biosynthesis from the natural precursor nicotinamide. Interference with this enzyme does not primarily intoxicate cells because the mitochondrial respiratory activity and the NAD+ -dependent redox reactions involved remain unaffected as long as NAD+ is not effectively depleted by catabolic reactions. Certain tissues, however, have a high turnover of NAD+ through its cleavage by enzymes like poly(ADP-ribose) polymerase. Such cells often rely on the more readily available nicotinamide pathway for NAD+ synthesis and undergo apoptosis after inhibition of NAPRT, whereas cells effectively using the nicotinic acid pathway for NAD+ synthesis remain unaffected. In support of this concept, FK866 effectively induced delayed cell death by apoptosis in HepG2 human liver carcinoma cells with an IC(50) of approximately 1 nM, did not directly inhibit mitochondrial respiratory activity, but caused gradual NAD+ depletion through specific inhibition of NAPRT. This enzyme, when partially purified from K562 human leukemia cells, was noncompetitively inhibited by FK866, and the inhibitor constants were calculated to be 0.4 nM for the enzyme/substrate complex (K(i)) and 0.3 nM for the free enzyme (K(i)'), respectively. Nicotinic acid and nicotinamide were both found to have antidote potential for the cellular effects of FK866. FK866 may be used for treatment of diseases implicating deregulated apoptosis such as cancer for immunosuppression or as a sensitizer for genotoxic agents. Furthermore, it may provide an important tool for investigation of the molecular triggers of the mitochondrial pathway leading to apoptosis through enabling temporal separation of NAD+ decrease from ATP breakdown and apoptosis by several days.