
DBZ
Notch pathway inhibitor; Inhibits γ-secretase

概要
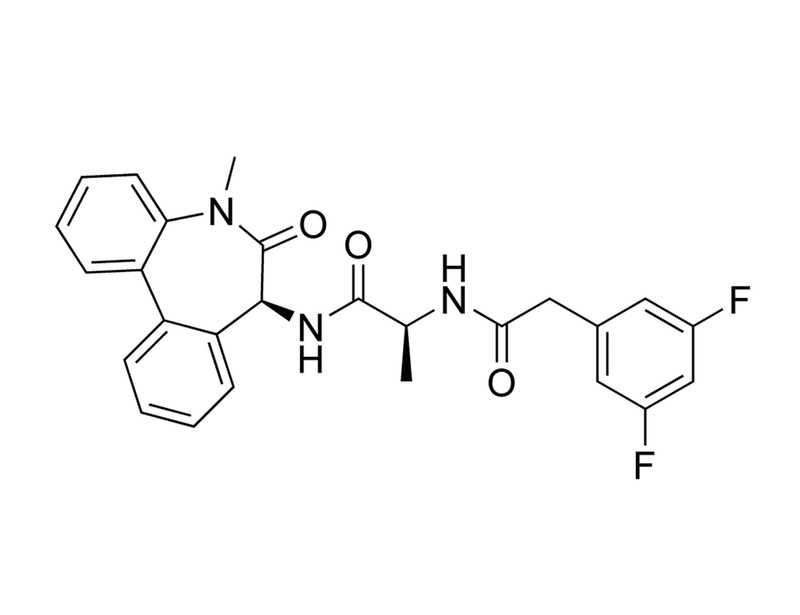
DBZ is a diazepine inhibitor of γ-secretase, which cleaves transmembrane proteins including Notch, amyloid precursor protein (APP), and Ephrin-B2 (Borgegard et al). DBZ blocks the cleavage of Notch into its active signalling effector, Notch intracellular domain, with an IC₅₀ of 1.7 nM (Milano et al.).
REPROGRAMMING
· Enables reprogramming of human keratinocytes to induced pluripotent stem cells in the absence of oncogenic reprogramming factors KLF4 and c-MYC (Ichida et al.).
DIFFERENTIATION
· Induces intestinal cell apoptosis & goblet cell metaplasia in rats; attenuates the reduction of paneth cells and goblet cells caused by tuberous sclerosis 2 (TSC2) inhibition (Milano et al.; Zhou et al.).
METABOLISM
· Improves glucose homeostasis and mediates a metabolic shift toward the utilization of fat as the energy source in mice (Bi et al.).
CANCER RESEARCH
· Induces differentiation of intestinal adenomas in Apc(Min) transgenic mice (van Es et al.).
· Decreases the production of inflammatory cytokines by alloreactive T cells after bone marrow transplantation in mice, reducing the severity of graft-versus-host disease (Tran et al.).
REPROGRAMMING
· Enables reprogramming of human keratinocytes to induced pluripotent stem cells in the absence of oncogenic reprogramming factors KLF4 and c-MYC (Ichida et al.).
DIFFERENTIATION
· Induces intestinal cell apoptosis & goblet cell metaplasia in rats; attenuates the reduction of paneth cells and goblet cells caused by tuberous sclerosis 2 (TSC2) inhibition (Milano et al.; Zhou et al.).
METABOLISM
· Improves glucose homeostasis and mediates a metabolic shift toward the utilization of fat as the energy source in mice (Bi et al.).
CANCER RESEARCH
· Induces differentiation of intestinal adenomas in Apc(Min) transgenic mice (van Es et al.).
· Decreases the production of inflammatory cytokines by alloreactive T cells after bone marrow transplantation in mice, reducing the severity of graft-versus-host disease (Tran et al.).
Alternative Names
Dibenzazepine, YO-01027
Cell Type
Cancer Cells and Cell Lines, Intestinal Cells, Pluripotent Stem Cells, T Cells
Species
Human, Mouse, Rat, Non-Human Primate, Other
Application
Reprogramming
Area of Interest
Cancer Research, Epithelial Cell Biology, Immunology, Metabolism, Stem Cell Biology, Transplantation Research
CAS Number
209984-56-5
Chemical Formula
C₂₆H₂₃F₂N₃O₃
Molecular Weight
463.5 g/mol
Purity
≥ 98%
Pathway
Notch
Target
γ-Secretase
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | DBZ | 73092 | All | English |
| Safety Data Sheet | DBZ | 73092 | All | English |
数据及文献
Publications (9)
Cell death & disease 2015 JAN
TSC2/mTORC1 signaling controls Paneth and goblet cell differentiation in the intestinal epithelium.
Abstract
Abstract
The intestinal mucosa undergoes a continual process of proliferation, differentiation and apoptosis, which is regulated by multiple signaling pathways. Notch signaling is critical for the control of intestinal stem cell maintenance and differentiation. However, the precise mechanisms involved in the regulation of differentiation are not fully understood. Previously, we have shown that tuberous sclerosis 2 (TSC2) positively regulates the expression of the goblet cell differentiation marker, MUC2, in intestinal cells. Using transgenic mice constitutively expressing a dominant negative TSC2 allele, we observed that TSC2 inactivation increased mTORC1 and Notch activities, and altered differentiation throughout the intestinal epithelium, with a marked decrease in the goblet and Paneth cell lineages. Conversely, treatment of mice with either Notch inhibitor dibenzazepine (DBZ) or mTORC1 inhibitor rapamycin significantly attenuated the reduction of goblet and Paneth cells. Accordingly, knockdown of TSC2 activated, whereas knockdown of mTOR or treatment with rapamycin decreased, the activity of Notch signaling in the intestinal cell line LS174T. Importantly, our findings demonstrate that TSC2/mTORC1 signaling contributes to the maintenance of intestinal epithelium homeostasis by regulating Notch activity.
Nature medicine 2014 AUG
Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity.
Abstract
Abstract
Beige adipocytes in white adipose tissue (WAT) are similar to classical brown adipocytes in that they can burn lipids to produce heat. Thus, an increase in beige adipocyte content in WAT browning would raise energy expenditure and reduce adiposity. Here we report that adipose-specific inactivation of Notch1 or its signaling mediator Rbpj in mice results in browning of WAT and elevated expression of uncoupling protein 1 (Ucp1), a key regulator of thermogenesis. Consequently, as compared to wild-type mice, Notch mutants exhibit elevated energy expenditure, better glucose tolerance and improved insulin sensitivity and are more resistant to high fat diet-induced obesity. By contrast, adipose-specific activation of Notch1 leads to the opposite phenotypes. At the molecular level, constitutive activation of Notch signaling inhibits, whereas Notch inhibition induces, Ppargc1a and Prdm16 transcription in white adipocytes. Notably, pharmacological inhibition of Notch signaling in obese mice ameliorates obesity, reduces blood glucose and increases Ucp1 expression in white fat. Therefore, Notch signaling may be therapeutically targeted to treat obesity and type 2 diabetes.
Nature chemical biology 2014 AUG
Notch inhibition allows oncogene-independent generation of iPS cells.
Abstract
Abstract
The reprogramming of somatic cells to pluripotency using defined transcription factors holds great promise for biomedicine. However, human reprogramming remains inefficient and relies either on the use of the potentially dangerous oncogenes KLF4 and CMYC or the genetic inhibition of the tumor suppressor gene p53. We hypothesized that inhibition of signal transduction pathways that promote differentiation of the target somatic cells during development might relieve the requirement for non-core pluripotency factors during induced pluripotent stem cell (iPSC) reprogramming. Here, we show that inhibition of Notch greatly improves the efficiency of iPSC generation from mouse and human keratinocytes by suppressing p21 in a p53-independent manner and thereby enriching for undifferentiated cells capable of long-term self-renewal. Pharmacological inhibition of Notch enabled routine production of human iPSCs without KLF4 and CMYC while leaving p53 activity intact. Thus, restricting the development of somatic cells by altering intercellular communication enables the production of safer human iPSCs.
The international journal of biochemistry & cell biology 2014
The Notch γ-secretase inhibitor ameliorates kidney fibrosis via inhibition of TGF-β/Smad2/3 signaling pathway activation.
Abstract
Abstract
Kidney fibrosis is a common feature of chronic kidney disease (CKD). A recent study suggests that abnormal Notch signaling activation contributes to the development of renal fibrosis. However, the molecular mechanism that regulates this process remains unexplored. Unilateral ureteral obstruction (UUO) or sham-operated C57BL6 mice (aged 10 weeks) were randomly assigned to receive dibenzazepine (DBZ, 250 μg/100g/d) or vehicle for 7 days. Histologic examinations were performed on the kidneys using Masson's trichrome staining and immunohistochemistry. Real-time PCR and western blot analysis were used for detection of mRNA expression and protein phosphorylation. The expression of Notch 1, 3, and 4, Notch intracellular domain (NICD), and its target genes Hes1 and HeyL were upregulated in UUO mice, while the increase in NICD protein was significantly attenuated by DBZ. After 7 days, the severity of renal fibrosis and expression of fibrotic markers, including collagen 1α1/3α1, fibronectin, and α-smooth muscle actin, were markedly increased in UUO compared with sham mice. In contrast, administration of DBZ markedly attenuated these effects. Furthermore, DBZ significantly inhibited UUO-induced expression of transforming growth factor (TGF)-β, phosphorylated Smad 2, and Smad 3. Mechanistically, Notch signaling activation in tubular epithelial cells enhanced fibroblast proliferation and activation in a coculture experiment. Our study provides evidence that Notch signaling is implicated in renal fibrogenesis. The Notch inhibitor DBZ can ameliorate this process via inhibition of the TGF-β/Smad2/3 signaling pathway, and might be a novel drug for preventing chronic kidney disease.
The Journal of clinical investigation 2013 APR
Blockade of individual Notch ligands and receptors controls graft-versus-host disease.
Abstract
Abstract
Graft-versus-host disease (GVHD) is the main complication of allogeneic bone marrow transplantation. Current strategies to control GVHD rely on global immunosuppression. These strategies are incompletely effective and decrease the anticancer activity of the allogeneic graft. We previously identified Notch signaling in T cells as a new therapeutic target for preventing GVHD. Notch-deprived T cells showed markedly decreased production of inflammatory cytokines, but normal in vivo proliferation, increased accumulation of regulatory T cells, and preserved anticancer effects. Here, we report that γ-secretase inhibitors can block all Notch signals in alloreactive T cells, but lead to severe on-target intestinal toxicity. Using newly developed humanized antibodies and conditional genetic models, we demonstrate that Notch1/Notch2 receptors and the Notch ligands Delta-like1/4 mediate all the effects of Notch signaling in T cells during GVHD, with dominant roles for Notch1 and Delta-like4. Notch1 inhibition controlled GVHD, but led to treatment-limiting toxicity. In contrast, Delta-like1/4 inhibition blocked GVHD without limiting adverse effects while preserving substantial anticancer activity. Transient blockade in the peritransplant period provided durable protection. These findings open new perspectives for selective and safe targeting of individual Notch pathway components in GVHD and other T cell-mediated human disorders.
The Journal of biological chemistry 2012 APR
First and second generation γ-secretase modulators (GSMs) modulate amyloid-β (Aβ) peptide production through different mechanisms.
Abstract
Abstract
γ-Secretase-mediated cleavage of amyloid precursor protein (APP) results in the production of Alzheimer disease-related amyloid-β (Aβ) peptides. The Aβ42 peptide in particular plays a pivotal role in Alzheimer disease pathogenesis and represents a major drug target. Several γ-secretase modulators (GSMs), such as the nonsteroidal anti-inflammatory drugs (R)-flurbiprofen and sulindac sulfide, have been suggested to modulate the Alzheimer-related Aβ production by targeting the APP. Here, we describe novel GSMs that are selective for Aβ modulation and do not impair processing of Notch, EphB2, or EphA4. The GSMs modulate Aβ both in cell and cell-free systems as well as lower amyloidogenic Aβ42 levels in the mouse brain. Both radioligand binding and cellular cross-competition experiments reveal a competitive relationship between the AstraZeneca (AZ) GSMs and the established second generation GSM, E2012, but a noncompetitive interaction between AZ GSMs and the first generation GSMs (R)-flurbiprofen and sulindac sulfide. The binding of a (3)H-labeled AZ GSM analog does not co-localize with APP but overlaps anatomically with a γ-secretase targeting inhibitor in rodent brains. Combined, these data provide compelling evidence of a growing class of in vivo active GSMs, which are selective for Aβ modulation and have a different mechanism of action compared with the original class of GSMs described.