
AKT Inhibitor X
PI3K/AKT pathway inhibitor; Inhibits AKT

概要
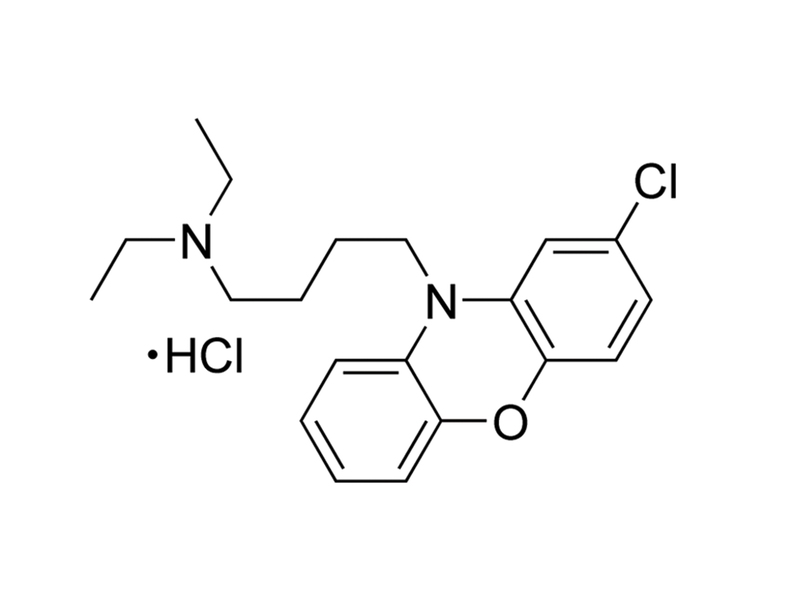
AKT Inhibitor X is a cell-permeable phenoxazine-derivative inhibitor of AKT kinase phosphorylation with an IC₅₀ of ~1 - 2 μM. AKT Inhibitor X blocks translocation of AKT after insulin-like growth factor 1 (IGF-1) treatment (Thimmaiah et al.). This product is supplied as the hydrochloride salt of the molecule.
CANCER RESEARCH
· Inhibits growth and induces apoptosis of human rhabdomyosarcoma cell lines (Thimmaiah et al.).
· Inhibits proliferation of breast cancer cell lines, alone or synergistically with chloroquine (Hu et al.).
· Reduces replication of Myxoma virus in a variety of human tumor cell lines (Werden & McFadden).
DISEASE MODELING
· Induces autophagy in neurons and is neuroprotective in a primary neuronal Huntington Disease cellular model (Tsvetkov et al.).
CANCER RESEARCH
· Inhibits growth and induces apoptosis of human rhabdomyosarcoma cell lines (Thimmaiah et al.).
· Inhibits proliferation of breast cancer cell lines, alone or synergistically with chloroquine (Hu et al.).
· Reduces replication of Myxoma virus in a variety of human tumor cell lines (Werden & McFadden).
DISEASE MODELING
· Induces autophagy in neurons and is neuroprotective in a primary neuronal Huntington Disease cellular model (Tsvetkov et al.).
Alternative Names
10-DEBC hydrochloride
Cell Type
Cancer Cells and Cell Lines, Mammary Cells, Neurons
Species
Human, Mouse, Rat, Non-Human Primate, Other
Area of Interest
Cancer Research, Disease Modeling, Neuroscience
CAS Number
925681-41-0
Chemical Formula
C₂₀H₂₅ClN₂O · HCl
Molecular Weight
381.3 g/mol
Purity
≥ 95%
Pathway
PI3K/AKT
Target
AKT
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | AKT Inhibitor X (Hydrochloride) | 72952 | All | English |
| Safety Data Sheet | AKT Inhibitor X (Hydrochloride) | 72952 | All | English |
数据及文献
Publications (4)
Journal of virology 2010
Pharmacological manipulation of the akt signaling pathway regulates myxoma virus replication and tropism in human cancer cells.
Abstract
Abstract
Viruses have evolved an assortment of mechanisms for regulating the Akt signaling pathway to establish a cellular environment more favorable for viral replication. Myxoma virus (MYXV) is a rabbit-specific poxvirus that encodes many immunomodulatory factors, including an ankyrin repeat-containing host range protein termed M-T5 that functions to regulate tropism of MYXV for rabbit lymphocytes and certain human cancer cells. MYXV permissiveness in these human cancer cells is dependent upon the direct interaction between M-T5 and Akt, which has been shown to induce the kinase activity of Akt. In this study, an array of compounds that selectively manipulate Akt signaling was screened and we show that only a subset of Akt inhibitors significantly decreased the ability of MYXV to replicate in previously permissive human cancer cells. Furthermore, reduced viral replication efficiency was correlated with lower levels of phosphorylated Akt. In contrast, the PP2A-specific phosphatase inhibitor okadaic acid promoted increased Akt kinase activation and rescued MYXV replication in human cancer cells that did not previously support viral replication. Finally, phosphorylation of Akt at residue Thr308 was shown to dictate the physical interaction between Akt and M-T5, which then leads to phosphorylation of Ser473 and permits productive MYXV replication in these human cancer cells. The results of this study further characterize the mechanism by which M-T5 exploits the Akt signaling cascade and affirms this interaction as a major tropism determinant that regulates the replication efficiency of MYXV in human cancer cells.
Proceedings of the National Academy of Sciences of the United States of America 2010
A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model.
Abstract
Abstract
Autophagy is an intracellular turnover pathway. It has special relevance for neurodegenerative proteinopathies, such as Alzheimer disease, Parkinson disease, and Huntington disease (HD), which are characterized by the accumulation of misfolded proteins. Although induction of autophagy enhances clearance of misfolded protein and has therefore been suggested as a therapy for proteinopathies, neurons appear to be less responsive to classic autophagy inducers than nonneuronal cells. Searching for improved inducers of neuronal autophagy, we discovered an N(10)-substituted phenoxazine that, at proper doses, potently and safely up-regulated autophagy in neurons in an Akt- and mTOR-independent fashion. In a neuron model of HD, this compound was neuroprotective and decreased the accumulation of diffuse and aggregated misfolded protein. A structure/activity analysis with structurally similar compounds approved by the US Food and Drug Administration revealed a defined pharmacophore for inducing neuronal autophagy. This pharmacophore should prove useful in studying autophagy in neurons and in developing therapies for neurodegenerative proteinopathies.
Bioorganic & medicinal chemistry 2008
The efficacy and selectivity of tumor cell killing by Akt inhibitors are substantially increased by chloroquine.
Abstract
Abstract
This study was to evaluate the enhancement value of chloroquine (CQ) in cancer cell killing when used in combination with Akt inhibitors. The results showed that the combination of CQ and Akt inhibitors is much more effective than either one alone. Importantly, the CQ-mediated chemosensitization of cell killing effects by Akt inhibitors is cancer specific. In particular, when combined with 10 microM CQ, 1,3-dihydro-1-(1-((4-(6-phenyl-1H-imidazo[4,5-g]quinoxalin-7-yl)phenyl)methyl)-4-piperidinyl)-2H-benzimidazol-2-one (an Akt1 and 2 inhibitor; compound 8) killed cancer cells 10-120 times more effectively than normal cells. Thus, CQ is a very effective and cancer-specific chemosensitizer when used in combination with Akt inhibitors.
The Journal of biological chemistry 2005 SEP
Identification of N10-substituted phenoxazines as potent and specific inhibitors of Akt signaling.
Abstract
Abstract
A series of 30 N10-substituted phenoxazines were synthesized and screened as potential inhibitors of Akt. In cellular assays at 5 mum, 17 compounds inhibited insulin-like growth factor 1 (IGF-I)-stimulated phosphorylation of Akt (Ser-473) by at least 50% but did not inhibit IGF-I-stimulated phosphorylation of Erk-1/2 (Thr-202/Tyr-204). Substitutions at the 2-position (Cl or CF3) did not alter inhibitory activity, whereas N10-substitutions with derivatives having acetyl (20B) or morpholino (12B) side chain lost activity compared with propyl or butyl substituents (7B and 14B). Inhibition of Akt phosphorylation was associated with the inhibition of IGF-I stimulation of the mammalian target of rapamycin phosphorylation (Ser-2448 and Ser-2481), phosphorylation of p70 S6 kinase (Thr-389), and ribosomal protein S6 (Ser-235/236) in Rh1, Rh18, and Rh30 cell lines. The two most potent compounds 10-[4'-(N-diethylamino)butyl]-2-chlorophenoxazine (10B) and 10-[4'-[(beta-hydroxyethyl)piperazino]butyl]-2-chlorophenoxazine (15B) (in vitro, IC50 approximately 1-2 microM) were studied further. Inhibition of Akt phosphorylation correlated with inhibition of its kinase activity as determined in vitro after immunoprecipitation. Akt inhibitory phenoxazines did not inhibit the activity of recombinant phosphatidylinositol 3'-kinase, PDK1, or SGK1 but potently inhibited the kinase activity of recombinant Akt and Akt deltaPH, a mutant lacking the pleckstrin homology domain. Akt inhibitory phenoxazines blocked IGF-I-stimulated nuclear translocation of Akt in Rh1 cells and suppressed growth of Rh1, Rh18, and Rh30 cells (IC50 2-5 microM), whereas inactive" derivatives were textgreater or = 10-fold less potent inhibitors of cell growth. In contrast to rapamycin analogs�