
AKT Inhibitor VIII
PI3K/AKT pathway inhibitor; Inhibits AKT1, AKT2, AKT3


概要
AKT Inhibitor VIII is a cell-permeable, allosteric inhibitor of all three forms of the kinase AKT (AKT1, AKT2, and AKT3) with IC₅₀ values of 58, 210, and 2,200 nM, respectively (Lindsley et al.; Calleja et al.). It displays good selectivity against a panel of 70 other kinases with micromolar inhibition against some kinases, such as calcium/calmodulin-dependent protein kinase 1 and smooth muscle myosin light-chain kinase (Logie et al.).
CANCER RESEARCH
· Sensitizes prostate tumor and cervical carcinoma cells to apoptotic stimuli (DeFeo-Jones et al.).
· Blocks mitosis and inhibits migration of HeLa cells (Jo et al.).
METABOLISM
· Reduces insulin-dependent gene repression in liver cells leading to reduced insulin sensitivity (Logie et al.).
CANCER RESEARCH
· Sensitizes prostate tumor and cervical carcinoma cells to apoptotic stimuli (DeFeo-Jones et al.).
· Blocks mitosis and inhibits migration of HeLa cells (Jo et al.).
METABOLISM
· Reduces insulin-dependent gene repression in liver cells leading to reduced insulin sensitivity (Logie et al.).
Alternative Names
AKTi-1/2; AKT 1/2 inhibitor
Cell Type
Cancer Cells and Cell Lines, Hepatic Cells
Species
Human, Mouse, Rat, Non-Human Primate, Other
Area of Interest
Cancer Research, Disease Modeling
CAS Number
612847-09-3
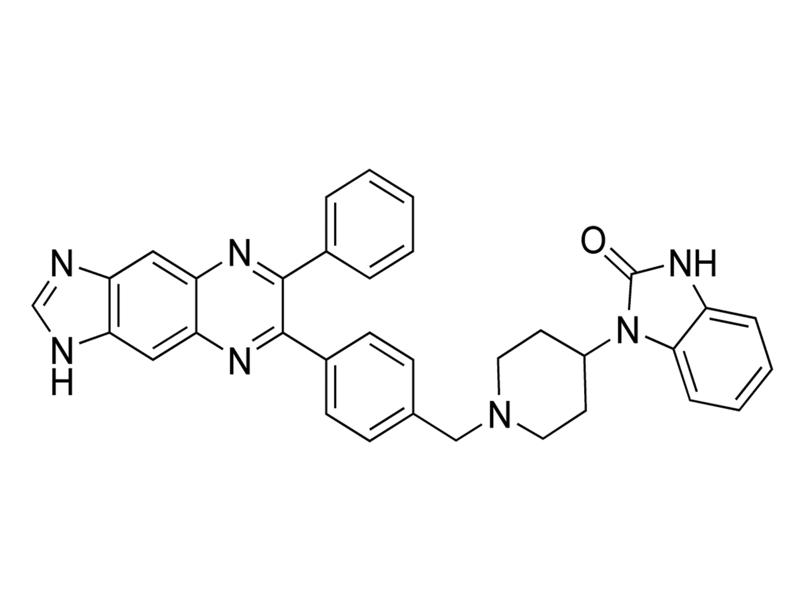
Chemical Formula
C₃₄H₂₉N₇O
Molecular Weight
551.7 g/mol
Purity
≥ 98%
Pathway
PI3K/AKT
Target
AKT1, AKT2, AKT3
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | AKT Inhibitor VIII | 72942, 72944 | All | English |
| Safety Data Sheet | AKT Inhibitor VIII | 72942, 72944 | All | English |
数据及文献
Publications (5)
Proceedings of the National Academy of Sciences of the United States of America 2011
Deactivation of Akt by a small molecule inhibitor targeting pleckstrin homology domain and facilitating Akt ubiquitination.
Abstract
Abstract
The phosphatidylinositol-3,4,5-triphosphate (PIP3) binding function of pleckstrin homology (PH) domain is essential for the activation of oncogenic Akt/PKB kinase. Following the PIP3-mediated activation at the membrane, the activated Akt is subjected to other regulatory events, including ubiquitination-mediated deactivation. Here, by identifying and characterizing an allosteric inhibitor, SC66, we show that the facilitated ubiquitination effectively terminates Akt signaling. Mechanistically, SC66 manifests a dual inhibitory activity that directly interferes with the PH domain binding to PIP3 and facilitates Akt ubiquitination. A known PH domain-dependent allosteric inhibitor, which stabilizes Akt, prevents the SC66-induced Akt ubiquitination. A cancer-relevant Akt1 (e17k) mutant is unstable, making it intrinsically sensitive to functional inhibition by SC66 in cellular contexts in which the PI3K inhibition has little inhibitory effect. As a result of its dual inhibitory activity, SC66 manifests a more effective growth suppression of transformed cells that contain a high level of Akt signaling, compared with other inhibitors of PIP3/Akt pathway. Finally, we show the anticancer activity of SC66 by using a soft agar assay as well as a mouse xenograft tumor model. In conclusion, in this study, we not only identify a dual-function Akt inhibitor, but also demonstrate that Akt ubiquitination could be chemically exploited to effectively facilitate its deactivation, thus identifying an avenue for pharmacological intervention in Akt signaling.
PLoS biology 2009
Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition.
Abstract
Abstract
Protein kinase B (PKB/Akt) belongs to the AGC superfamily of related serine/threonine protein kinases. It is a key regulator downstream of various growth factors and hormones and is involved in malignant transformation and chemo-resistance. Full-length PKB protein has not been crystallised, thus studying the molecular mechanisms that are involved in its regulation in relation to its structure have not been simple. Recently, the dynamics between the inactive and active conformer at the molecular level have been described. The maintenance of PKB's inactive state via the interaction of the PH and kinase domains prevents its activation loop to be phosphorylated by its upstream activator, phosphoinositide-dependent protein kinase-1 (PDK1). By using a multidisciplinary approach including molecular modelling, classical biochemical assays, and Förster resonance energy transfer (FRET)/two-photon fluorescence lifetime imaging microscopy (FLIM), a detailed model depicting the interaction between the different domains of PKB in its inactive conformation was demonstrated. These findings in turn clarified the molecular mechanism of PKB inhibition by AKT inhibitor VIII (a specific allosteric inhibitor) and illustrated at the molecular level its selectivity towards different PKB isoforms. Furthermore, these findings allude to the possible function of the C-terminus in sustaining the inactive conformer of PKB. This study presents essential insights into the quaternary structure of PKB in its inactive conformation. An understanding of PKB structure in relation to its function is critical for elucidating its mode of activation and discovering how to modulate its activity. The molecular mechanism of inhibition of PKB activation by the specific drug AKT inhibitor VIII has critical implications for determining the mechanism of inhibition of other allosteric inhibitors and for opening up opportunities for the design of new generations of modulator drugs.
Diabetes 2007
Characterization of a protein kinase B inhibitor in vitro and in insulin-treated liver cells.
Abstract
Abstract
OBJECTIVE: Abnormal expression of the hepatic gluconeogenic genes (glucose-6-phosphatase [G6Pase] and PEPCK) contributes to hyperglycemia. These genes are repressed by insulin, but this process is defective in diabetic subjects. Protein kinase B (PKB) is implicated in this action of insulin. An inhibitor of PKB, Akt inhibitor (Akti)-1/2, was recently reported; however, the specificity and efficacy against insulin-induced PKB was not reported. Our aim was to characterize the specificity and efficacy of Akti-1/2 in cells exposed to insulin and then establish whether inhibition of PKB is sufficient to prevent regulation of hepatic gene expression by insulin. RESEARCH DESIGN AND METHODS: Akti-1/2 was assayed against 70 kinases in vitro and its ability to block PKB activation in cells exposed to insulin fully characterized. RESULTS: Akti-1/2 exhibits high selectivity toward PKBalpha and PKBbeta. Complete inhibition of PKB activity is achieved in liver cells incubated with 1-10 mumol/l Akti-1/2, and this blocks insulin regulation of PEPCK and G6Pase expression. Our data demonstrate that only 5-10% of maximal insulin-induced PKB is required to fully repress PEPCK and G6Pase expression. Finally, we demonstrate reduced insulin sensitivity of these gene promoters in cells exposed to submaximal concentrations of Akti-1/2; however, full repression of the genes can still be achieved by high concentrations of insulin. CONCLUSIONS: This work establishes the requirement for PKB activity in the insulin regulation of PEPCK, G6Pase, and a third insulin-regulated gene, IGF-binding protein-1 (IGFBP1); suggests a high degree of functional reserve; and identifies Akti-1/2 as a useful tool to delineate PKB function in the liver.
Molecular cancer therapeutics 2005 FEB
Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members.
Abstract
Abstract
Recent studies indicate that dysregulation of the Akt/PKB family of serine/threonine kinases is a prominent feature of many human cancers. The Akt/PKB family is composed of three members termed Akt1/PKBalpha, Akt2/PKBbeta, and Akt3/PKBgamma. It is currently not known to what extent there is functional overlap between these family members. We have recently identified small molecule inhibitors of Akt. These compounds have pleckstrin homology domain-dependent, isozyme-specific activity. In this report, we present data showing the relative contribution that inhibition of the different isozymes has on the apoptotic response of tumor cells to a variety of chemotherapies. In multiple cell backgrounds, maximal induction of caspase-3 activity is achieved when both Akt1 and Akt2 are inhibited. This induction is not reversed by overexpression of functionally active Akt3. The level of caspase-3 activation achieved under these conditions is equivalent to that observed with the phosphatidylinositol-3-kinase inhibitor LY294002. We also show that in different tumor cell backgrounds inhibition of mammalian target of rapamycin, a downstream substrate of Akt, is less effective in inducing caspase-3 activity than inhibition of Akt1 and Akt2. This shows that the survival phenotype conferred by Akt can be mediated by signaling pathways independent of mammalian target of rapamycin in some tumor cell backgrounds. Finally, we show that inhibition of both Akt1 and Akt2 selectively sensitizes tumor cells, but not normal cells, to apoptotic stimuli.
Bioorganic & medicinal chemistry letters 2005
Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors.
Abstract
Abstract
This letter describes the development of two series of potent and selective allosteric Akt kinase inhibitors that display an unprecedented level of selectivity for either Akt1, Akt2 or both Akt1/Akt2. An iterative analog library synthesis approach quickly provided a highly selective Akt1/Akt2 inhibitor that induces apoptosis in tumor cells and inhibits Akt phosphorylation in vivo.