
WHI-P131
JAK/STAT pathway inhibitor; Inhibits JAK3



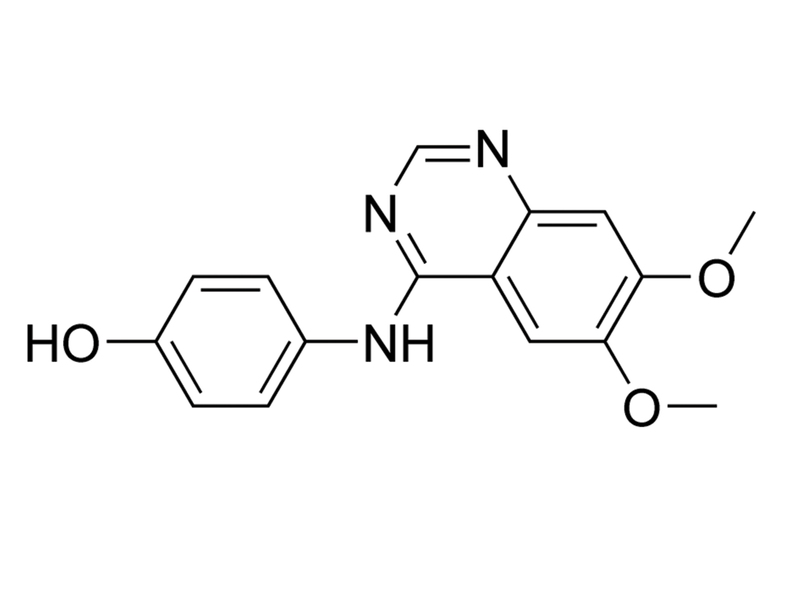
概要
WHI-P131 is an inhibitor of Janus kinase 3 (JAK3) with IC₅₀ values of 9 and 78 μM against human and mouse proteins, respectively (Sudbeck et al.). It has also been reported to show significant inhibition of other kinases, including epidermal growth factor receptor (EGFR) in the nanomolar range (Changelian et al.; Uckun et al.). No significant inhibition of JAK1 or JAK2 has been observed (Sudbeck et al.).
CANCER RESEARCH
· Induces apoptosis and cell death in human glioblastoma cell lines U373 and U87 (Narla et al.).
IMMUNOLOGY
· Inhibits lipopolysaccharide (LPS)-induced nitric oxide synthase expression and nitric oxide production in macrophages (Sareila et al.).
MAINTENANCE
· Inhibits proliferation of neural stem cell-containing neurospheres at lower concentrations than the concentration required to inhibit proliferation of astrocyte cultures (Diamandis et al.).
CANCER RESEARCH
· Induces apoptosis and cell death in human glioblastoma cell lines U373 and U87 (Narla et al.).
IMMUNOLOGY
· Inhibits lipopolysaccharide (LPS)-induced nitric oxide synthase expression and nitric oxide production in macrophages (Sareila et al.).
MAINTENANCE
· Inhibits proliferation of neural stem cell-containing neurospheres at lower concentrations than the concentration required to inhibit proliferation of astrocyte cultures (Diamandis et al.).
Alternative Names
Janex-1
Cell Type
Cancer Cells and Cell Lines, Monocytes, Neural Stem and Progenitor Cells
Species
Human, Mouse, Rat, Non-Human Primate, Other
Area of Interest
Cancer Research, Immunology
CAS Number
202475-60-3
Chemical Formula
C₁₆H₁₅N₃O₃
Molecular Weight
297.3 g/mol
Purity
≥ 98%
Pathway
JAK/STAT
Target
JAK3
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | WHI-P131 | 73542, 73544 | All | English |
| Safety Data Sheet | WHI-P131 | 73542, 73544 | All | English |
数据及文献
Publications (6)
International immunopharmacology 2008
Janus kinase 3 inhibitor WHI-P154 in macrophages activated by bacterial endotoxin: differential effects on the expression of iNOS, COX-2 and TNF-alpha.
Abstract
Abstract
Bacterial endotoxin is a potent inducer of inflammatory response, including the induction of inducible nitric oxide synthase (iNOS) expression and nitric oxide (NO) production, and the expression of cyclo-oxygenase (COX)-2 and tumor necrosis factor (TNF)-alpha in inflammatory cells. In the present study, we investigated the effects of pharmacological inhibition of Janus kinase (JAK) 3 on the production of these proinflammatory molecules in macrophages exposed to bacterial endotoxin (lipopolysaccharide; LPS). JAK3 inhibitors WHI-P154 (4-(3'-bromo-4'-hydroxylphenyl)-amino-6,7-dimethoxyquinazoline) and its derivative WHI-P131 inhibited LPS-induced iNOS expression and NO production in a dose-dependent manner. WHI-P154 inhibited the activation of signal transducer and activator of transcription (STAT) 1 and the expression of iNOS mRNA but it had no effect on iNOS mRNA decay when determined by actinomycin D assay. The JAK3 inhibitor had no effect on COX-2 expression, and TNF-alpha production was slightly inhibited only at higher drug concentrations (30 microM). In addition, WHI-P154 inhibited iNOS expression and NO production also in human epithelial cells. Our results suggest that JAK3 inhibition modulates human and murine iNOS expression and NO production in response to inflammatory stimuli.
Blood 2008
The specificity of JAK3 kinase inhibitors.
Abstract
Abstract
PF-956980 is a selective inhibitor of JAK3, related in structure to CP-690550, a compound being evaluated in clinical trials for rheumatoid arthritis and prevention of allograft rejection. PF-956980 has been evaluated against a panel of 30 kinases, and found to have nanomolar potency against only JAK3. Cellular and whole blood activity of this compound parallels its potency and selectivity in enzyme assays. It was effective in vivo at inhibiting the delayed type hypersensivity reaction in mice. We compared 2 commercially available JAK3 inhibitors (WHI-P131 and WHI-P154) in the same panel of biochemical and cellular assays and found them to be neither potent nor selective for JAK3. Both were found to be nanomolar inhibitors of the EGF receptor family of kinases. As these compounds have been used in numerous publications in the transplant and autoimmune disease literature, their specificity should be considered when interpreting these results.
Nature chemical biology 2007
Chemical genetics reveals a complex functional ground state of neural stem cells.
Abstract
Abstract
The identification of self-renewing and multipotent neural stem cells (NSCs) in the mammalian brain holds promise for the treatment of neurological diseases and has yielded new insight into brain cancer. However, the complete repertoire of signaling pathways that governs the proliferation and self-renewal of NSCs, which we refer to as the 'ground state', remains largely uncharacterized. Although the candidate gene approach has uncovered vital pathways in NSC biology, so far only a few highly studied pathways have been investigated. Based on the intimate relationship between NSC self-renewal and neurosphere proliferation, we undertook a chemical genetic screen for inhibitors of neurosphere proliferation in order to probe the operational circuitry of the NSC. The screen recovered small molecules known to affect neurotransmission pathways previously thought to operate primarily in the mature central nervous system; these compounds also had potent inhibitory effects on cultures enriched for brain cancer stem cells. These results suggest that clinically approved neuromodulators may remodel the mature central nervous system and find application in the treatment of brain cancer.
Current cancer drug targets 2001
Structure-based design of novel anticancer agents.
Abstract
Abstract
Recently identified agents that interact with cytoskeletal elements such as tubulin include synthetic spiroketal pyrans (SPIKET) and monotetrahydrofuran compounds (COBRA compounds). SPIKET compounds target the spongistatin binding site of beta-tubulin and COBRA compounds target a unique binding cavity on alpha-tubulin. At nanomolar concentrations, the SPIKET compound SPIKET-P causes tubulin depolymerization and exhibits potent cytotoxic activity against cancer cells. COBRA-1 inhibits GTP-induced tubulin polymerization. Treatment of human breast cancer and brain tumor cells with COBRA-1 caused destruction of microtubule organization and apoptosis. Other studies have identified some promising protein tyrosine kinase inhibitors as anti-cancer agents. These include EGFR inhibitors such as the quinazoline derivative WHI-P97 and the leflunomide metabolite analog LFM-A12. Both LFM-A12 and WHI-P97 inhibit the in vitro invasiveness of EGFR positive human breast cancer cells at micromolar concentrations and induce apoptotic cell death. Dimethoxyquinazoline compounds WHI-P131 and WHI-P154 inhibit tyrosine kinase JAK3 in leukemia cells. Of particular interest is WHI-P131, which inhibits JAK3 but not JAK1, JAK2, SYK, BTK, LYN, or IRK at concentrations as high as 350 microM. Studies of BTK inhibitors showed that the leflunomide metabolite analog LFM-A13 inhibited BTK in leukemia and lymphoma cells. Consistent with the anti-apoptotic function of BTK, treatment of leukemic cells with LFM-A13 enhanced their sensitivity to chemotherapy-induced apoptosis.
Clinical cancer research : an official journal of the American Association for Cancer Research 1999
Structure-based design of specific inhibitors of Janus kinase 3 as apoptosis-inducing antileukemic agents.
Abstract
Abstract
A novel homology model of the kinase domain of Janus kinase (JAK) 3 was used for the structure-based design of dimethoxyquinazoline compounds with potent and specific inhibitory activity against JAK3. The active site of JAK3 in this homology model measures roughly 8 A x 11 A x 20 A, with a volume of approximately 530 A3 available for inhibitor binding. Modeling studies indicated that 4-(phenyl)-amino-6,7-dimethoxyquinazoline (parent compound WHI-258) would likely fit into the catalytic site of JAK3 and that derivatives of this compound that contain an OH group at the 4' position of the phenyl ring would more strongly bind to JAK3 because of added interactions with Asp-967, a key residue in the catalytic site of JAK3. These predictions were consistent with docking studies indicating that compounds containing a 4'-OH group, WHI-P131 [4-(4'-hydroxyphenyl)-amino-6,7-dimethoxyquinazoline], WHI-P154 [4-(3'-bromo-4'-hydroxylphenyl)-amino-6,7-dimethoxyquinazoline], and WHI-P97 [4-(3',5'-dibromo-4'-hydroxylphenyl)-amino-6,7-dimethoxyquinazolin e], were likely to bind favorably to JAK3, with estimated K(i)s ranging from 0.6 to 2.3 microM. These compounds inhibited JAK3 in immune complex kinase assays in a dose-dependent fashion. In contrast, compounds lacking the 4'-OH group, WHI-P79 [4-(3'-bromophenyl)-amino-6,7-dimethoxyquinazoline], WHI-P111 [4-(3'-bromo-4'-methylphenyl)-amino-6,7-dimethoxyquinazoline], WHI-P112 [4-(2',5'-dibromophenyl)-amino-6,7-dimethoxyquinazoline], WHI-P132 [4-(2'-hydroxylphenyl)-amino-6,7-dimethoxyquinazoline], and WHI-P258 [4-(phenyl)-amino-6,7-dimethoxyquinazoline], were predicted to bind less strongly, with estimated K(i)s ranging from 28 to 72 microM. These compounds did not show any significant JAK3 inhibition in kinase assays. Furthermore, the lead dimethoxyquinazoline compound, WHI-P131, which showed potent JAK3-inhibitory activity (IC50 of 78 microM), did not inhibit JAK1 and JAK2, the ZAP/SYK family tyrosine kinase SYK, the TEC family tyrosine kinase BTK, the SRC family tyrosine kinase LYN, or the receptor family tyrosine kinase insulin receptor kinase, even at concentrations as high as 350 microM. WHI-P131 induced apoptosis in JAK3-expressing human leukemia cell lines NALM-6 and LC1;19 but not in melanoma (M24-MET) or squamous carcinoma (SQ20B) cells. Leukemia cells were not killed by dimethoxyquinazoline compounds that were inactive against JAK3. WHI-P131 inhibited the clonogenic growth of JAK3-positive leukemia cell lines DAUDI, RAMOS, LC1;19, NALM-6, MOLT-3, and HL-60 (but not JAK3-negative BT-20 breast cancer, M24-MET melanoma, or SQ20B squamous carcinoma cell lines) in a concentration-dependent fashion. Potent and specific inhibitors of JAK3 such as WHI-P131 may provide the basis for the design of new treatment strategies against acute lymphoblastic leukemia, the most common form of childhood cancer.
Clinical cancer research : an official journal of the American Association for Cancer Research 1998
4-(3'-Bromo-4'hydroxylphenyl)-amino-6,7-dimethoxyquinazoline: a novel quinazoline derivative with potent cytotoxic activity against human glioblastoma cells.
Abstract
Abstract
The novel quinazoline derivative 4-(3'-bromo-4'-hydroxylphenyl)-amino-6,7-dimethoxyquinazoline (WHI-P154) exhibited significant cytotoxicity against U373 and U87 human glioblastoma cell lines, causing apoptotic cell death at micromolar concentrations. The in vitro antiglioblastoma activity of WHI-P154 was amplified textgreater 200-fold and rendered selective by conjugation to recombinant human epidermal growth factor (EGF). The EGF-P154 conjugate was able to bind to and enter target glioblastoma cells within 10-30 min via receptor (R)-mediated endocytosis by inducing internalization of the EGF-R molecules. In vitro treatment with EGF-P154 resulted in killing of glioblastoma cells at nanomolar concentrations with an IC50 of 813 +/- 139 nM, whereas no cytotoxicity against EGF-R-negative leukemia cells was observed, even at concentrations as high as 100 microM. The in vivo administration of EGF-P154 resulted in delayed tumor progression and improved tumor-free survival in a severe combined immunodeficient mouse glioblastoma xenograft model. Whereas none of the control mice remained alive tumor-free beyond 33 days (median tumor-free survival, 19 days) and all control mice had tumors that rapidly progressed to reach an average size of textgreater 500 mm3 by 58 days, 40% of mice treated for 10 consecutive days with 1 mg/kg/day EGF-P154 remained alive and free of detectable tumors for more than 58 days with a median tumor-free survival of 40 days. The tumors developing in the remaining 60% of the mice never reached a size textgreater 50 mm3. Thus, targeting WHI-P154 to the EGF-R may be useful in the treatment of glioblastoma multiforme.