
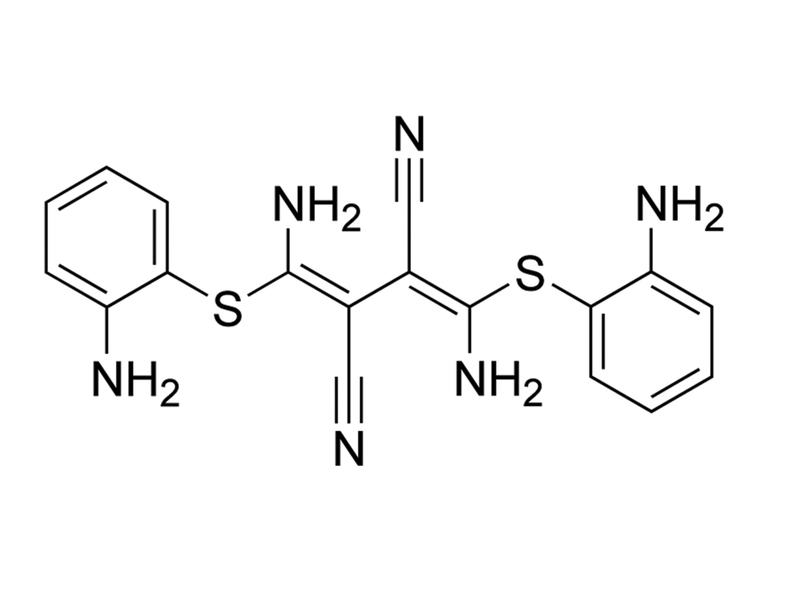
U-0126
MEK/ERK pathway inhibitor; Inhibits MEK1 and MEK2


概要
U-0126 is a selective, non-ATP competitive inhibitor of mitogen activated protein kinase kinase (MEK), inhibiting MEK1 and MEK2 with IC₅₀ values of 72 nM and 58 nM, respectively (Favata et al.; Scherle et al.). It shows little or no inhibition at micromolar levels of other kinases such as ERK, protein kinase C (PKC), c-Jun N-terminal kinases (JNK), other MAP kinase kinases (MKK3, MKK4, MKK6), cyclin-dependent kinases (CDK2, CDK4), ABL, and RAF (Favata et al. 1998). U-0126 also antagonizes AP-1 transcription and selectively inhibits promoters containing AP-1 response elements (Favata et al.).
MAINTENANCE & SELF-RENEWAL
· In combination with basic FGF, Activin A, and a PKC inhibitor, U-0126 promotes maintenance of human pluripotent stem cells (Kinehara et al.)
· Used alone with MEF-conditioned medium, U-0126 inhibits self-renewal of human pluripotent stem cells, leading to differentiation, without affecting proliferation or survival (Li et al.).
· Inhibits glutamate-induced oxidative stress in mouse hippocampal HT22 cells and rat primary cortical cultures (Satoh et al.; Ong et al.).
· Neuroprotective in a gerbil ischemia model and in primary mouse neurons cultured under hypoxia (Namura et al.).
IMMUNOLOGY
· Inhibits T-cell proliferation against certain antigenic stimuli by reducing IL-2 mRNA levels (DeSilva et al.).
DISEASE MODELING
· Activates peroxisome proliferator activated receptor (PPAR) co-activator 1α (PGC-1α) and prevents neurotoxicity and spatial memory impairment in rats challenged by amyloid beta (Aβ; Ashabi et al.).
MAINTENANCE & SELF-RENEWAL
· In combination with basic FGF, Activin A, and a PKC inhibitor, U-0126 promotes maintenance of human pluripotent stem cells (Kinehara et al.)
· Used alone with MEF-conditioned medium, U-0126 inhibits self-renewal of human pluripotent stem cells, leading to differentiation, without affecting proliferation or survival (Li et al.).
· Inhibits glutamate-induced oxidative stress in mouse hippocampal HT22 cells and rat primary cortical cultures (Satoh et al.; Ong et al.).
· Neuroprotective in a gerbil ischemia model and in primary mouse neurons cultured under hypoxia (Namura et al.).
IMMUNOLOGY
· Inhibits T-cell proliferation against certain antigenic stimuli by reducing IL-2 mRNA levels (DeSilva et al.).
DISEASE MODELING
· Activates peroxisome proliferator activated receptor (PPAR) co-activator 1α (PGC-1α) and prevents neurotoxicity and spatial memory impairment in rats challenged by amyloid beta (Aβ; Ashabi et al.).
Alternative Names
Not applicable
Cell Type
Neurons, Pluripotent Stem Cells, T Cells
Species
Human, Mouse, Rat, Non-Human Primate, Other
Application
Maintenance
Area of Interest
Disease Modeling, Immunology, Neuroscience, Stem Cell Biology
CAS Number
109511-58-2
Chemical Formula
C₁₈H₁₆N₆S₂
Molecular Weight
380.5 g/mol
Purity
≥ 98%
Pathway
MEK/ERK
Target
MEK1, MEK2
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | U-0126 | 73522, 73524 | All | English |
| Safety Data Sheet | U-0126 | 73522, 73524 | All | English |
数据及文献
Publications (9)
ACS chemical neuroscience 2015
U0126 protects cells against oxidative stress independent of its function as a MEK inhibitor.
Abstract
Abstract
U0126 is a potent and selective inhibitor of MEK1 and MEK2 kinases. It has been widely used as an inhibitor for the Ras/Raf/MEK/ERK signaling pathway with over 5000 references on the NCBI PubMed database. In particular, U0126 has been used in a number of studies to show that inhibition of the Raf/MEK/ERK pathway protects neuronal cells against oxidative stress. Here, we report that U0126 can function as an antioxidant that protects PC12 cells against a number of different oxidative-stress inducers. This protective effect of U0126 is independent of its function as a MEK inhibitor, as several other MEK inhibitors failed to show similar protective effects. U0126 reduces reactive oxygen species (ROS) in cells. We further demonstrate that U0126 is a direct ROS scavenger in vitro, and the oxidation products of U0126 exhibit fluorescence. Our finding that U0126 is a strong antioxidant signals caution for its future usage as a MEK inhibitor and for interpreting some previous results.
PloS one 2013
Protein kinase C regulates human pluripotent stem cell self-renewal.
Abstract
Abstract
BACKGROUND: The self-renewal of human pluripotent stem (hPS) cells including embryonic stem and induced pluripotent stem cells have been reported to be supported by various signal pathways. Among them, fibroblast growth factor-2 (FGF-2) appears indispensable to maintain self-renewal of hPS cells. However, downstream signaling of FGF-2 has not yet been clearly understood in hPS cells. METHODOLOGY/PRINCIPAL FINDINGS: In this study, we screened a kinase inhibitor library using a high-throughput alkaline phosphatase (ALP) activity-based assay in a minimal growth factor-defined medium to understand FGF-2-related molecular mechanisms regulating self-renewal of hPS cells. We found that in the presence of FGF-2, an inhibitor of protein kinase C (PKC), GF109203X (GFX), increased ALP activity. GFX inhibited FGF-2-induced phosphorylation of glycogen synthase kinase-3β (GSK-3β), suggesting that FGF-2 induced PKC and then PKC inhibited the activity of GSK-3β. Addition of activin A increased phosphorylation of GSK-3β and extracellular signal-regulated kinase-1/2 (ERK-1/2) synergistically with FGF-2 whereas activin A alone did not. GFX negated differentiation of hPS cells induced by the PKC activator, phorbol 12-myristate 13-acetate whereas Gö6976, a selective inhibitor of PKCα, β, and γ isoforms could not counteract the effect of PMA. Intriguingly, functional gene analysis by RNA interference revealed that the phosphorylation of GSK-3β was reduced by siRNA of PKCδ, PKCε, and ζ, the phosphorylation of ERK-1/2 was reduced by siRNA of PKCε and ζ, and the phosphorylation of AKT was reduced by PKCε in hPS cells. CONCLUSIONS/SIGNIFICANCE: Our study suggested complicated cross-talk in hPS cells that FGF-2 induced the phosphorylation of phosphatidylinositol-3 kinase (PI3K)/AKT, mitogen-activated protein kinase/ERK-1/2 kinase (MEK), PKC/ERK-1/2 kinase, and PKC/GSK-3β. Addition of GFX with a MEK inhibitor, U0126, in the presence of FGF-2 and activin A provided a long-term stable undifferentiated state of hPS cells even though hPS cells were dissociated into single cells for passage. This study untangles the cross-talk between molecular mechanisms regulating self-renewal and differentiation of hPS cells.
Behavioural brain research 2012
ERK and p38 inhibitors attenuate memory deficits and increase CREB phosphorylation and PGC-1α levels in Aβ-injected rats.
Abstract
Abstract
In this study, we investigated the effect of intracerebroventricular administration of ERK and p38 specific inhibitors, U0126 and PD169316, respectively, on learning and memory deficits induced by amyloid beta (Aβ) in rats. To investigate the effects of these compounds on learning and memory, we performed Morris water maze (MWM) test. U0126 and/or PD169316 improved spatial learning in MWM in Aβ-injected rats, 20 days after Aβ-injection. To determine the mechanisms of action of U0126 and PD169316, we studies their effect on some intracellular signaling pathways such as Ca(+)/cAMP-response element binding protein (CREB), c-fos, and transcription factors that regulate mitochondrial biogenesis. Based on our data, CREB and c-fos levels decreased 7 days after Aβ-injection, while U0126 and/or PD169316 pretreatments significantly increased these levels. Moreover, U0126 and PD169316 activated peroxisome proliferator-activated receptor gamma coactivator-1a, nuclear respiratory factor 1, and mitochondrial transcription factor A, 7 days after Aβ-injection. Surprisingly, these factors were returned to vehicle level, 20 days after Aβ-injection. Our findings reinforce the potential neuroprotective effect of these inhibitors against the Aβ toxicity.
Differentiation; research in biological diversity 2007
MEK/ERK signaling contributes to the maintenance of human embryonic stem cell self-renewal.
Abstract
Abstract
MEK/ERK signaling plays a crucial role in a diverse set of cellular functions including cell proliferation, differentiation and survival, and recently has been reported to negatively regulate mouse embryonic stem cell (mESC) self-renewal by antagonizing STAT3 activity. However, its role in human ESCs (hESCs) remains unclear. Here we investigated the functions of MEK/ERK in controlling hESC activity. We demonstrated that MEK/ERK kinases were targets of fibroblast growth factor (FGF) pathway in hESCs. Surprisingly, we found that, in contrast to mESCs, high basal MEK/ERK activity was required for maintaining hESCs in an undifferentiated state. Inhibition of MEK/ERK activity by specific MEK inhibitors PD98059 and U0126, or by RNA interference, rapidly caused the loss of self-renewal capacity. We also showed that MEK/ERK signaling cooperated with phosphoinositide 3-kinase (PI3K)/AKT signaling in maintaining hESC pluripotency. However, MEK/ERK signaling had little or no effect on regulating hESC proliferation and survival, in contrast to PI3K/AKT signaling. Taken together, these findings reveal the unique and crucial role of MEK/ERK signaling in the determination of hESC cell fate and expand our understanding of the molecular mechanisms behind the FGF pathway maintenance of hESC pluripotency. Importantly, these data make evident the striking differences in the control of self-renewal between hESCs and mESCs.
Proceedings of the National Academy of Sciences of the United States of America 2001
Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia.
Abstract
Abstract
Brain subjected to acute ischemic attack caused by an arterial blockage needs immediate arterial recanalization. However, restoration of cerebral blood flow can cause tissue injury, which is termed reperfusion injury. It is important to inhibit reperfusion injury to achieve greater brain protection. Because oxidative stress has been shown to activate mitogen-activated protein kinases (MAPKs), and because oxidative stress contributes to reperfusion injury, MAPK may be a potential target to inhibit reperfusion injury after brain ischemia. Here, we demonstrate that reperfusion after forebrain ischemia dramatically increases phosphorylation level of extracellular signal-regulated kinase 2 (ERK2) in the gerbil hippocampus. In addition, i.v. administration of U0126 (100-200 mg/kg), a specific inhibitor of MEK (MAPK/ERK kinase), protects the hippocampus against forebrain ischemia. Moreover, treatment with U0126 at 3 h after ischemia significantly reduces infarct volume after transient (3 h) focal cerebral ischemia in mice. This protection is accompanied by reduced phosphorylation level of ERK2, substrates for MEK, in the damaged brain areas. Furthermore, U0126 protects mouse primary cultured cortical neurons against oxygen deprivation for 9 h as well as nitric oxide toxicity. These results provide further evidence for the role of MEK/ERK activation in brain injury resulting from ischemia/reperfusion, and indicate that MEK inhibition may increase the resistance of tissue to ischemic injury.
Neuroscience letters 2000
Neuroprotection by MAPK/ERK kinase inhibition with U0126 against oxidative stress in a mouse neuronal cell line and rat primary cultured cortical neurons.
Abstract
Abstract
Oxidative stress is implicated in the pathogenesis of neuronal degenerative diseases. Oxidative stress has been shown to activate extracellular signal-regulated kinases (ERK)1/2. We investigated the role of these mitogen-activated protein kinases (MAPKs) in oxidative neuronal injury by using a mouse hippocampal cell line (HT22) and rat primary cortical cultures. Here, we show that a novel MAPK/ERK kinase (MEK) specific inhibitor U0126 profoundly protected HT22 cells against oxidative stress induced by glutamate, which was accompanied by an inhibition of phosphorylation of ERK1/2. U0126 also protected rat primary cultured cortical neurons against glutamate or hypoxia. However, U0126 was not protective against death caused by tumor necrosis factor alpha (TNFalpha), A23187, or staurosporine. These results indicate that MEK plays a central role in the neuronal death caused by oxidative stress.