
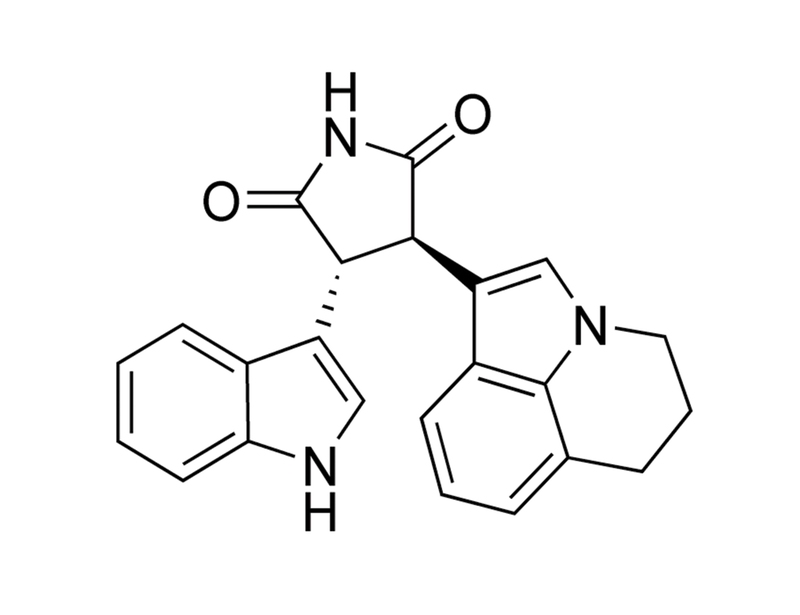
Tivantinib
HGF pathway inhibitor; Inhibits MET


概要
Tivantinib is a staurosporine derivative which binds to inactive, dephosphorylated MET receptor tyrosine kinase, in a manner that is not competitive with ATP (Munshi et al.; Eathiraj et al.). It is selective for MET (Ki ≅ 355 nM) in a screen of 230 kinases (Munshi et al.). It also shows cytotoxic activity which is distinct from its inhibition of MET (Basilico et al.; Katayama et al.). It has also been shown to bind directly to the colchicine binding pocket of tubulin, thereby reducing tubulin polymerization (Aoyama et al.).
CANCER RESEARCH
· Induces apoptosis of human myeloma CD138+ plasma cells in vitro, and demonstrates efficacy in a mouse xenograft model of myeloma (Zaman et al.).
· Inhibits metastatic growth of breast cancer cells in bone and reduces tumor-induced osteolysis, in a mouse xenograft model (Previdi et al.).
· Perturbs microtubule dynamics, induces G2/M arrest, and promotes apoptosis independently of c-MET inhibition in a variety of cancer cell lines (Basilico et al.).
CANCER RESEARCH
· Induces apoptosis of human myeloma CD138+ plasma cells in vitro, and demonstrates efficacy in a mouse xenograft model of myeloma (Zaman et al.).
· Inhibits metastatic growth of breast cancer cells in bone and reduces tumor-induced osteolysis, in a mouse xenograft model (Previdi et al.).
· Perturbs microtubule dynamics, induces G2/M arrest, and promotes apoptosis independently of c-MET inhibition in a variety of cancer cell lines (Basilico et al.).
Alternative Names
ARQ 197
Cell Type
Cancer Cells and Cell Lines
Species
Human, Mouse, Rat, Non-Human Primate, Other
Area of Interest
Cancer Research
CAS Number
905854-02-6
Chemical Formula
C₂₃H₁₉N₃O₂
Molecular Weight
369.4 g/mol
Purity
≥ 98%
Pathway
HGF
Target
MET
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | Tivantinib | 73482, 73484 | All | English |
| Safety Data Sheet | Tivantinib | 73482, 73484 | All | English |
数据及文献
Publications (7)
Neoplasia (New York, N.Y.) 2015
Targeting the pro-survival protein MET with tivantinib (ARQ 197) inhibits growth of multiple myeloma cells.
Abstract
Abstract
The hepatocyte growth factor (HGF)/MNNG HOS transforming gene (MET) pathway regulates cell growth, survival, and migration. MET is mutated or amplified in several malignancies. In myeloma, MET is not mutated, but patients have high plasma concentrations of HGF, high levels of MET expression, and gene copy number, which are associated with poor prognosis and advanced disease. Our previous studies demonstrated that MET is critical for myeloma cell survival and its knockdown induces apoptosis. In our current study, we tested tivantinib (ARQ 197), a small-molecule pharmacological MET inhibitor. At clinically achievable concentrations, tivantinib induced apoptosis by textgreater50% in all 12 human myeloma cell lines tested. This biologic response was associated with down-regulation of MET signaling and inhibition of the mitogen-activated protein kinase and phosphoinositide 3-kinase pathways, which are downstream of the HGF/MET axis. Tivantinib was equally effective in inducing apoptosis in myeloma cell lines resistant to standard chemotherapy (melphalan, dexamethasone, bortezomib, and lenalidomide) as well as in cells that were co-cultured with a protective bone marrow microenvironment or with exogenous cytokines. Tivantinib induced apoptosis in CD138+ plasma cells from patients and demonstrated efficacy in a myeloma xenograft mouse model. On the basis of these data, we initiated a clinical trial for relapsed/refractory multiple myeloma (MM). In conclusion, MET inhibitors may be an attractive target-based strategy for the treatment of MM.
Molecular cancer therapeutics 2014
Tivantinib (ARQ 197) exhibits antitumor activity by directly interacting with tubulin and overcomes ABC transporter-mediated drug resistance.
Abstract
Abstract
Tivantinib (ARQ197) was first reported as a highly selective inhibitor of c-MET and is currently being investigated in a phase III clinical trial. However, as recently reported by us and another group, tivantinib showed cytotoxic activity independent of cellular c-MET status and also disrupted microtubule dynamics. To investigate if tivantinib exerts its cytotoxic activity by disrupting microtubules, we quantified polymerized tubulin in cells and xenograft tumors after tivantinib treatment. Consistent with our previous report, tivantinib reduced tubulin polymerization in cells and in mouse xenograft tumors in vivo. To determine if tivantinib directly binds to tubulin, we performed an in vitro competition assay. Tivantinib competitively inhibited colchicine but not vincristine or vinblastine binding to purified tubulin. These results imply that tivantinib directly binds to the colchicine binding site of tubulin. To predict the binding mode of tivantinib with tubulin, we performed computer simulation of the docking pose of tivantinib with tubulin using GOLD docking program. Computer simulation predicts tivantinib fitted into the colchicine binding pocket of tubulin without steric hindrance. Furthermore, tivantinib showed similar IC50 values against parental and multidrug-resistant cells. In contrast, other microtubule-targeting drugs, such as vincristine, paclitaxel, and colchicine, could not suppress the growth of cells overexpressing ABC transporters. Moreover, the expression level of ABC transporters did not correlate with the apoptosis-inducing ability of tivantinib different from other microtubule inhibitor. These results suggest that tivantinib can overcome ABC transporter-mediated multidrug-resistant tumor cells and is potentially useful against various tumors.
Clinical Cancer Research 2013
Tivantinib (ARQ197) Displays Cytotoxic Activity That Is Independent of Its Ability to Bind MET
Abstract
Abstract
PURPOSE: MET, the high-affinity receptor for hepatocyte growth factor, is frequently deregulated in human cancer. Tivantinib (ARQ197; Arqule), a staurosporine derivative that binds to the dephosphorylated MET kinase in vitro, is being tested clinically as a highly selective MET inhibitor. However, the mechanism of action of tivantinib is still unclear. EXPERIMENTAL DESIGN: The activity of tivantinib was analyzed in multiple cellular models, including: cells displaying c-MET gene amplification, strictly 'addicted' to MET signaling; cells with normal c-MET gene copy number, not dependent on MET for growth; cells not expressing MET; somatic knockout cells in which the ATP-binding cleft of MET, where tivantinib binds, was deleted by homologous recombination; and a cell system 'poisoned' by MET kinase hyperactivation, where cells die unless cultured in the presence of a specific MET inhibitor. RESULTS: Tivantinib displayed cytotoxic activity independently of c-MET gene copy number and regardless of the presence or absence of MET. In both wild-type and isogenic knockout cells, tivantinib perturbed microtubule dynamics, induced G2/M arrest, and promoted apoptosis. Tivantinib did not rescue survival of cells 'poisoned' by MET kinase hyperactivation, but further incremented cell death. In all cell models analyzed, tivantinib did not inhibit HGF-dependent or -independent MET tyrosine autophosphorylation. CONCLUSIONS: We conclude that tivantinib displays cytotoxic activity via molecular mechanisms that are independent from its ability to bind MET. This notion has a relevant impact on the interpretation of clinical results, on the design of future clinical trials, and on the selection of patients receiving tivantinib treatment.
Cancer research 2013
Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition.
Abstract
Abstract
The receptor tyrosine kinase c-MET is the high-affinity receptor for the hepatocyte growth factor (HGF). The HGF/c-MET axis is often dysregulated in tumors. c-MET activation can be caused by MET gene amplification, activating mutations, and auto- or paracrine mechanisms. Thus, c-MET inhibitors are under development as anticancer drugs. Tivantinib (ARQ 197) was reported as a small-molecule c-MET inhibitor and early clinical studies suggest antitumor activity. To assess whether the antitumor activity of tivantinib was due to inhibition of c-MET, we compared the activity of tivantinib with other c-MET inhibitors in both c-MET-addicted and nonaddicted cancer cells. As expected, other c-MET inhibitors, crizotinib and PHA-665752, suppressed the growth of c-MET-addicted cancers, but not the growth of cancers that are not addicted to c-MET. In contrast, tivantinib inhibited cell viability with similar potency in both c-MET-addicted and nonaddicted cells. These results suggest that tivantinib exhibits its antitumor activity in a manner independent of c-MET status. Tivantinib treatment induced a G(2)-M cell-cycle arrest in EBC1 cells similarly to vincristine treatment, whereas PHA-665752 or crizotinib treatment markedly induced G(0)-G(1) cell-cycle arrest. To identify the additional molecular target of tivantinib, we conducted COMPARE analysis, an in silico screening of a database of drug sensitivities across 39 cancer cell lines (JFCR39), and identified microtubule as a target of tivantinib. Tivantinib-treated cells showed typical microtubule disruption similar to vincristine and inhibited microtubule assembly in vitro. These results suggest that tivantinib inhibits microtubule polymerization in addition to inhibiting c-MET.
Molecular cancer therapeutics 2012
Breast cancer-derived bone metastasis can be effectively reduced through specific c-MET inhibitor tivantinib (ARQ 197) and shRNA c-MET knockdown.
Abstract
Abstract
Breast cancer exhibits a propensity to metastasize to bone, resulting in debilitating skeletal complications associated with significant morbidity and poor prognosis. The cross-talk between metastatic cancer cells and bone is critical to the development and progression of bone metastases. We have shown the involvement of the HGF/c-MET system in tumor-bone interaction contributing to human breast cancer metastasis. Therefore, disruption of HGF/c-MET signaling is a potential targeted approach to treating metastatic bone disease. In this study, we evaluated the effects of c-MET inhibition by both an oral, selective, small-molecule c-MET inhibitor, tivantinib, and a specific short hairpin RNA (shRNA) against c-MET in a mouse model of human breast cancer. Tivantinib exhibited dose-dependent antimetastatic activity in vivo, and the 120 mg/kg dose, proven to be suboptimal in reducing subcutaneous tumor growth, induced significant inhibition of metastatic growth of breast cancer cells in bone and a noteworthy reduction of tumor-induced osteolysis. shRNA-mediated c-MET silencing did not affect in vitro proliferation of bone metastatic cells, but significantly reduced their migration, and this effect was further enhanced by tivantinib. Both observations were confirmed in vivo. Indeed, more pronounced tumor growth suppression with concomitant marked decreases of lytic lesions and prolongation of survival were achieved by dual c-MET inhibition using both tivantinib and RNA interference strategies. Overall, our findings highlighted the effectiveness of c-MET inhibition in delaying the onset and progression of bone metastases and strongly suggest that targeting c-MET may have promising therapeutic value in the treatment of bone metastases from breast cancer.
The Journal of biological chemistry 2011
Discovery of a novel mode of protein kinase inhibition characterized by the mechanism of inhibition of human mesenchymal-epithelial transition factor (c-Met) protein autophosphorylation by ARQ 197.
Abstract
Abstract
A number of human malignancies exhibit sustained stimulation, mutation, or gene amplification of the receptor tyrosine kinase human mesenchymal-epithelial transition factor (c-Met). ARQ 197 is a clinically advanced, selective, orally bioavailable, and well tolerated c-Met inhibitor, currently in Phase 3 clinical testing in non-small cell lung cancer patients. Herein, we describe the molecular and structural basis by which ARQ 197 selectively targets c-Met. Through our analysis we reveal a previously undisclosed, novel inhibitory mechanism that utilizes distinct regulatory elements of the c-Met kinase. The structure of ARQ 197 in complex with the c-Met kinase domain shows that the inhibitor binds a conformation that is distinct from published kinase structures. ARQ 197 inhibits c-Met autophosphorylation and is highly selective for the inactive or unphosphorylated form of c-Met. Through our analysis of the interplay between the regulatory and catalytic residues of c-Met, and by comparison between the autoinhibited canonical conformation of c-Met bound by ARQ 197 to previously described kinase domains of type III receptor tyrosine kinases, we believe this to be the basis of a powerful new in silico approach for the design of similar inhibitors for other protein kinases of therapeutic interest.