
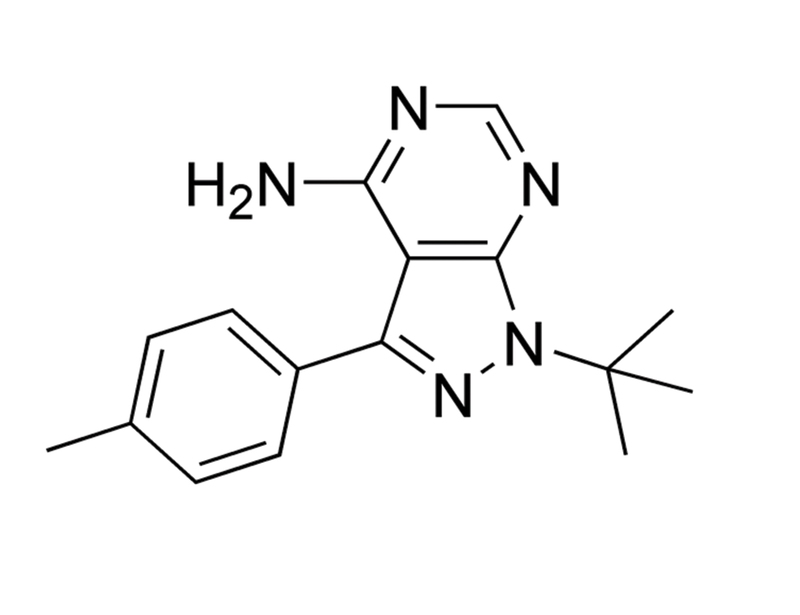
PP1
Tyrosine kinase inhibitor; Inhibits LCK, FYN, HCK, and SRC


概要
PP1 is a reversible inhibitor of the SRC family of tyrosine kinases. It inhibits LCK, FYN, HCK and SRC with IC₅₀ values of 5, 6, 20 and 170 nM, respectively (Hanke et al.). It is relatively selective for SRC family kinases versus other kinases, inhibiting epidermal growth factor receptor (EGFR), janus-activated kinase 2 (JAK2) and zeta-chain-associated protein kinase 70 (ZAP70) with IC₅₀ values of 0.25, > 50, and > 100 μM, respectively, and KIT, platelet-derived growth factor receptor (PDGFR), and RET tyrosine kinase in the 75 - 100 nM range (Carlomagno et al. ; Tatton et al.; Waltenberger et al.; Hanke et al.). PP1 also blocks TGF-β-mediated cellular responses by directly inhibiting type I TGF-β receptors (IC₅₀ = 50 nM; Ungefroren et al.; Maeda et al.).
REPROGRAMMING
· Enables reprogramming of mouse embryonic fibroblasts to induced pluripotent stem cells in the absence of reprogramming factor SOX2 (Staerk et al.; Ma et al.).
CANCER RESEARCH
· Blocks TGF-β-mediated migration of primary non-small cell lung carcinoma cells and pancreatic ductal adenocarcinoma cell lines (Bartscht et al.).
· Induces apoptosis in non-small cell lung cancer cell lines (Zhang et al.).
REPROGRAMMING
· Enables reprogramming of mouse embryonic fibroblasts to induced pluripotent stem cells in the absence of reprogramming factor SOX2 (Staerk et al.; Ma et al.).
CANCER RESEARCH
· Blocks TGF-β-mediated migration of primary non-small cell lung carcinoma cells and pancreatic ductal adenocarcinoma cell lines (Bartscht et al.).
· Induces apoptosis in non-small cell lung cancer cell lines (Zhang et al.).
Alternative Names
AGL 1872; EI 275
Cell Type
Cancer Cells and Cell Lines, Pluripotent Stem Cells
Species
Human, Mouse, Rat, Non-Human Primate, Other
Application
Reprogramming
Area of Interest
Cancer Research, Stem Cell Biology
CAS Number
172889-26-8
Chemical Formula
C₁₆H₁₉N₅
Molecular Weight
281.4 g/mol
Purity
≥ 98%
Pathway
Tyrosine Kinase
Target
LCK, SRC
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | PP1 | 73112, 73114 | All | English |
| Safety Data Sheet | PP1 | 73112, 73114 | All | English |
数据及文献
Publications (12)
Circulation research 2013
Progress in the reprogramming of somatic cells.
Abstract
Abstract
Pluripotent stem cells can differentiate into nearly all types of cells in the body. This unique potential provides significant promise for cell-based therapies to restore tissues or organs destroyed by injuries, degenerative diseases, aging, or cancer. The discovery of induced pluripotent stem cell (iPSC) technology offers a possible strategy to generate patient-specific pluripotent stem cells. However, because of concerns about the specificity, efficiency, kinetics, and safety of iPSC reprogramming, improvements or fundamental changes in this process are required before their effective clinical use. A chemical approach is regarded as a promising strategy to improve and change the iPSC process. Dozens of small molecules have been identified that can functionally replace reprogramming factors and significantly improve iPSC reprogramming. In addition to the prospect of deriving patient-specific tissues and organs from iPSCs, another attractive strategy for regenerative medicine is transdifferentiation-the direct conversion of one somatic cell type to another. Recent studies revealed a new paradigm of transdifferentiation: using transcription factors used in iPSC generation to induce transdifferentiation or called iPSC transcription factor-based transdifferentiation. This type of transdifferentiation not only reveals and uses the developmentally plastic intermediates generated during iPSC reprogramming but also produces a wide range of cells, including expandable tissue-specific precursor cells. Here, we review recent progress of small molecule approaches in the generation of iPSCs. In addition, we summarize the new concept of iPSC transcription factor-based transdifferentiation and discuss its application in generating various lineage-specific cells, especially cardiovascular cells.
Cancer chemotherapy and pharmacology 2012
The Src family kinase inhibitors PP2 and PP1 effectively block TGF-beta1-induced cell migration and invasion in both established and primary carcinoma cells.
Abstract
Abstract
PURPOSE: We have previously demonstrated that in pancreatic ductal adenocarcinoma (PDAC)-derived cell lines, the common Src family kinase inhibitors PP2 and PP1 effectively inhibited morphologic alterations associated with TGFβ1-mediated epithelial-to-mesenchymal transition (EMT) by blocking the kinase activity of the TGF-β type I receptor ALK5 rather than Src (Ungefroren et al. in Curr Cancer Drug Targets 11:524, 2011). In this report, the ability of PP2 and PP1, the more specific Src inhibitor SU6656, and the ALK5 inhibitor SB431542 to functionally block TGF-β1-dependent EMT and cell motility in established PDAC (Panc-1, Colo 357) and primary NSCLC (Tu459) cell lines were investigated. METHODS: The effects of PP2, PP1, SU6656, and SB431542 on TGF-β1-dependent cell scattering/EMT, cell migration/invasion, and expression of invasion-associated genes were measured by using the real-time cell analysis assay on the xCELLigence system and quantitative real-time RT-PCR, respectively. RESULTS: In all three cell lines tested, PP1, PP2, and SB431542 effectively blocked TGF-β1-induced cell scattering/EMT, migration, and invasion and in Colo 357 cells inhibited the induction of the invasion-associated MMP2 and MMP9 genes. In contrast, SU6656 only blocked TGF-β1-induced invasion in Panc-1 and Tu459 but not Colo 357 cells. PP1, and to a greater extent PP2, also inhibited the high spontaneous migratory activity of Panc-1 cells expressing a kinase-active ALK5 mutant. CONCLUSIONS: These data provide evidence that PP2 and PP1 are powerful inhibitors of TGF-β-induced cell migration and invasion in vitro and directly target ALK5. Both agents may be useful as dual TGF-β/Src inhibitors in experimental therapeutics to prevent metastatic spread in late-stage PDAC and NSCLC.
Current cancer drug targets 2011
The Src family kinase inhibitors PP2 and PP1 block TGF-beta1-mediated cellular responses by direct and differential inhibition of type I and type II TGF-beta receptors.
Abstract
Abstract
Both the nonreceptor tyrosine kinase Src and the receptors for transforming growth factor (TGF)-β (TβRI, TβRII) play major roles during tumorigenesis by regulating cell growth, migration/invasion and metastasis. The common Src family kinase inhibitors PP2 and PP1 effectively block Src activity in vitro and in vivo, however, they may exert non-specific effects on other kinases. In this study, we have evaluated PP2 and PP1 for their ability to inhibit TGFβ1-mediated responses in the TGF-β-responsive pancreatic adenocarcinoma cell line Panc1. We show that PP2 and PP1 but not the more specific Src inhibitor SU6656 effectively relieved TGF-b1-induced growth arrest and p21(WAF1) induction, while basal growth was enhanced by PP2 and PP1, and suppressed by SU6656. PP2 and PP1 but not SU6656 also suppressed TGF-β1-induced epithelial-to-mesenchymal transition (EMT) as evidenced by their ability to inhibit downregulation of the epithelial marker E-cadherin, and upregulation of the EMT-associated transcription factor Slug. Likewise, PP2 and PP1 but not SU6656 effectively blocked TGF-β1-induced activation of Smad2 and p38 MAPK and partially suppressed Smad activation and transcriptional activity on TGF-β/Smad-responsive reporters of a kinase-active TβRI mutant ectopically expressed in Panc1 cells. Interestingly, PP2 and PP1 strongly inhibited recombinant TβRI in an in vitro kinase assay, with PP1 being more potent and PP2 being nearly as potent as the established TβRI inhibitor SB431542. PP2 but not PP1 also weakly inhibited the TβRII kinase. Together, these data provide evidence that PP2 and PP1 are powerful inhibitors of TβR function that can block TGF-β/Smad signaling in a Src-unrelated fashion. Both agents may be useful as dual TGF-β/Src inhibitors in experimental therapeutics of late stage metastatic disease.
Angewandte Chemie (International ed. in English) 2011
Pan-Src family kinase inhibitors replace Sox2 during the direct reprogramming of somatic cells.
Abstract
Abstract
The Biochemical journal 2007 DEC
The selectivity of protein kinase inhibitors: a further update.
Abstract
Abstract
The specificities of 65 compounds reported to be relatively specific inhibitors of protein kinases have been profiled against a panel of 70-80 protein kinases. On the basis of this information, the effects of compounds that we have studied in cells and other data in the literature, we recommend the use of the following small-molecule inhibitors: SB 203580/SB202190 and BIRB 0796 to be used in parallel to assess the physiological roles of p38 MAPK (mitogen-activated protein kinase) isoforms, PI-103 and wortmannin to be used in parallel to inhibit phosphatidylinositol (phosphoinositide) 3-kinases, PP1 or PP2 to be used in parallel with Src-I1 (Src inhibitor-1) to inhibit Src family members; PD 184352 or PD 0325901 to inhibit MKK1 (MAPK kinase-1) or MKK1 plus MKK5, Akt-I-1/2 to inhibit the activation of PKB (protein kinase B/Akt), rapamycin to inhibit TORC1 [mTOR (mammalian target of rapamycin)-raptor (regulatory associated protein of mTOR) complex], CT 99021 to inhibit GSK3 (glycogen synthase kinase 3), BI-D1870 and SL0101 or FMK (fluoromethylketone) to be used in parallel to inhibit RSK (ribosomal S6 kinase), D4476 to inhibit CK1 (casein kinase 1), VX680 to inhibit Aurora kinases, and roscovitine as a pan-CDK (cyclin-dependent kinase) inhibitor. We have also identified harmine as a potent and specific inhibitor of DYRK1A (dual-specificity tyrosine-phosphorylated and -regulated kinase 1A) in vitro. The results have further emphasized the need for considerable caution in using small-molecule inhibitors of protein kinases to assess the physiological roles of these enzymes. Despite being used widely, many of the compounds that we analysed were too non-specific for useful conclusions to be made, other than to exclude the involvement of particular protein kinases in cellular processes.
The American journal of pathology 2007
SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines.
Abstract
Abstract
The role of Src-family kinases (SFKs) in non-small cell lung cancer (NSCLC) has not been fully defined. Here we addressed this question by examining SFK phosphorylation in NSCLC biopsy samples and using genetic and pharmacological approaches to inhibit SFK expression and activity in cultured NSCLC cells. Immunohistochemical analysis of NSCLC biopsy samples using a Tyr416 phosphorylation-specific, pan-SFK antibody revealed staining in 123 (33%) of 370 tumors. Because c-Src is known to be both an upstream activator and downstream mediator of epidermal growth factor receptor (EGFR), we next investigated SFK phosphorylation in a panel of NSCLC cell lines, including ones that depend on EGFR for survival. The EGFR-dependent NSCLC cell lines HCC827 and H3255 had increased phosphorylation of SFKs, and treatment of these cells with an SFK inhibitor (PP1 or SKI-606) induced apoptosis. PP1 decreased phosphorylation of EGFR, ErbB2, and ErbB3 and strikingly enhanced apoptosis by gefitinib, an EGFR inhibitor. HCC827 cells transfected with c-Src short hairpin RNA exhibited diminished phosphorylation of EGFR and ErbB2 and decreased sensitivity to apoptosis by PP1 or gefitinib. We conclude that SFKs are activated in NSCLC biopsy samples, promote the survival of EGFR-dependent NSCLC cells, and should be investigated as therapeutic targets in NSCLC patients.