
Nilotinib
Tyrosine kinase inhibitor; Inhibits BCR/ABL and ABL


概要
Nilotinib is a second-generation inhibitor of the oncogenic tyrosine kinase BCR-ABL with IC₅₀ values of 19, 140, and 9,200 nM for wild-type, E255K, and T315I mutant forms of BCR-ABL, respectively (Kitagawa et al.; Verstovsek et al.; O’Hare). It binds to the ATP-binding pocket of ABL, with higher affinity than imatinib (Manley et al. 2006; Verstovsek et al.). It also has activity below 1 μM against discoidin domain receptors (DDR) -1 and -2, platelet-derived growth factor receptors (PDGFR) –α and -β, stem cell factor receptor (KIT), and colony-stimulating factor 1 receptor (CSF-1R; Manley et al. 2010).
CANCER RESEARCH
· Inhibits cellular proliferation in many wild-type and mutant forms of Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) and chronic myeloid leukemia (CML) cells (Verstovsek et al.; O’Hare).
· Inhibits cell proliferation and progression through S phase in human lung cell line A549 through transcriptional changes in DNA helicase complex, cyclins, and cyclin-dependent kinases (Ji et al.).
CANCER RESEARCH
· Inhibits cellular proliferation in many wild-type and mutant forms of Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) and chronic myeloid leukemia (CML) cells (Verstovsek et al.; O’Hare).
· Inhibits cell proliferation and progression through S phase in human lung cell line A549 through transcriptional changes in DNA helicase complex, cyclins, and cyclin-dependent kinases (Ji et al.).
Alternative Names
AMN107; Tasigna
Cell Type
Cancer Cells and Cell Lines, Leukemia/Lymphoma Cells
Species
Human, Mouse, Rat, Non-Human Primate, Other
Area of Interest
Cancer Research
CAS Number
641571-10-0
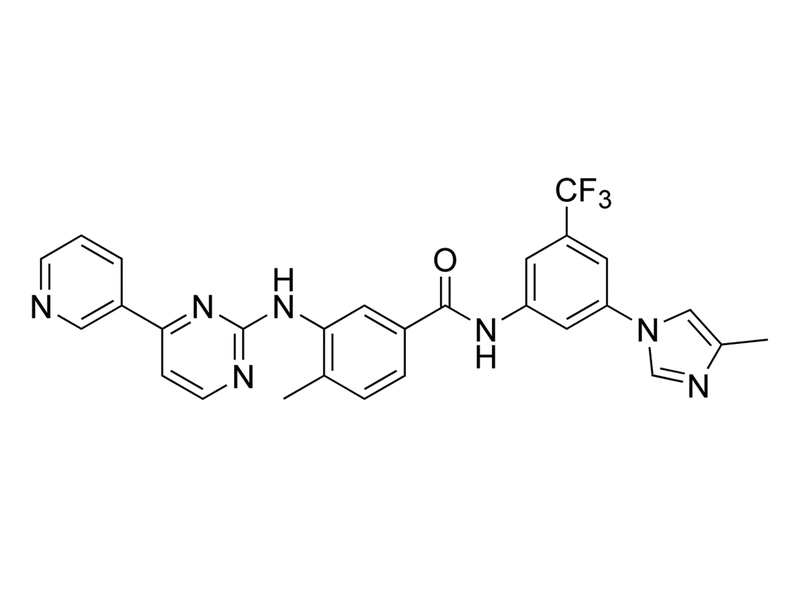
Chemical Formula
C₂₈H₂₂F₃N₇O
Molecular Weight
529.5 g/mol
Purity
≥ 95%
Pathway
Tyrosine Kinase
Target
ABL, BCR/ABL
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | Nilotinib | 73302, 73304 | All | English |
数据及文献
Publications (5)
Genes to cells : devoted to molecular & cellular mechanisms 2013
Activity-based kinase profiling of approved tyrosine kinase inhibitors.
Abstract
Abstract
The specificities of nine approved tyrosine kinase inhibitors (imatinib, dasatinib, nilotinib, gefitinib, erlotinib, lapatinib, sorafenib, sunitinib, and pazopanib) were determined by activity-based kinase profiling using a large panel of human recombinant active kinases. This panel consisted of 79 tyrosine kinases, 199 serine/threonine kinases, three lipid kinases, and 29 disease-relevant mutant kinases. Many potential targets of each inhibitor were identified by kinase profiling at the K(m) for ATP. In addition, profiling at a physiological ATP concentration (1 mm) was carried out, and the IC(50) values of the inhibitors against each kinase were compared with the estimated plasma-free concentration (calculated from published pharmacokinetic parameters of plasma C(trough) and C(max) values). This analysis revealed that the approved kinase inhibitors were well optimized for their target kinases. This profiling also implicates activity at particular off-target kinases in drug side effects. Thus, large-scale kinase profiling at both K(m) and physiological ATP concentrations could be useful in characterizing the targets and off-targets of kinase inhibitors.
Biochimica et biophysica acta 2010
Extended kinase profile and properties of the protein kinase inhibitor nilotinib.
Abstract
Abstract
As a drug used to treat imatinib-resistant and -intolerant, chronic and advanced phase chronic myelogenous leukaemia, nilotinib is well characterised as a potent inhibitor of the Abl tyrosine kinase activity of wild-type and imatinib-resistant mutant forms of BCR-Abl. Here we review the profile of nilotinib as a protein kinase inhibitor. Although an ATP-competitive inhibitor of Abl, nilotinib binds to a catalytically inactive conformation (DFG-out) of the activation loop. As a consequence of this, nilotinib exhibits time-dependent inhibition of Abl kinase in enzymatic assays, which can be extrapolated to other targets to explain differences between biochemical activity and cellular assays. Although these differences confound assessment of kinase selectivity, as assessed using a combination of protein binding and transphosphorylation assays, together with cellular autophosporylation and proliferation assays, well established kinase targets of nilotinib in rank order of inhibitory potency are DDR-1textgreaterDDR-2textgreaterBCR-Abl (Abl)textgreaterPDGFRalpha/betatextgreaterKITtextgreaterCSF-1R. In addition nilotinib has now been found to bind to both MAPK11 (p38beta) and MAPK12 (p38alpha), as well as with very high affinity to ZAK kinase. Although neither enzymatic nor cellular data are yet available to substantiate the drug as an inhibitor of ZAK phosphorylation, modeling predicts that it binds in an ATP-competitive fashion.
PLoS computational biology 2009 SEP
Transcriptional profiling of the dose response: a more powerful approach for characterizing drug activities.
Abstract
Abstract
The dose response curve is the gold standard for measuring the effect of a drug treatment, but is rarely used in genomic scale transcriptional profiling due to perceived obstacles of cost and analysis. One barrier to examining transcriptional dose responses is that existing methods for microarray data analysis can identify patterns, but provide no quantitative pharmacological information. We developed analytical methods that identify transcripts responsive to dose, calculate classical pharmacological parameters such as the EC50, and enable an in-depth analysis of coordinated dose-dependent treatment effects. The approach was applied to a transcriptional profiling study that evaluated four kinase inhibitors (imatinib, nilotinib, dasatinib and PD0325901) across a six-logarithm dose range, using 12 arrays per compound. The transcript responses proved a powerful means to characterize and compare the compounds: the distribution of EC50 values for the transcriptome was linked to specific targets, dose-dependent effects on cellular processes were identified using automated pathway analysis, and a connection was seen between EC50s in standard cellular assays and transcriptional EC50s. Our approach greatly enriches the information that can be obtained from standard transcriptional profiling technology. Moreover, these methods are automated, robust to non-optimized assays, and could be applied to other sources of quantitative data.
Cancer Research 2005
In vitro Activity of Bcr-Abl Inhibitors AMN107 and BMS-354825 against Clinically Relevant Imatinib-Resistant Abl Kinase Domain Mutants
Abstract
Abstract
Imatinib, a Bcr-Abl tyrosine kinase inhibitor, is a highly effective therapy for patients with chronic myelogenous leukemia (CML). Despite durable responses in most chronic phase patients, relapses have been observed and are much more prevalent in patients with advanced disease. The most common mechanism of acquired imatinib resistance has been traced to Bcr-Abl kinase domain mutations with decreased imatinib sensitivity. Thus, alternate Bcr-Abl kinase inhibitors that have activity against imatinib-resistant mutants would be useful for patients who relapse on imatinib therapy. Two such Bcr-Abl inhibitors are currently being evaluated in clinical trials: the improved potency, selective Abl inhibitor AMN107 and the highly potent dual Src/Abl inhibitor BMS-354825. In the current article, we compared imatinib, AMN107, and BMS-354825 in cellular and biochemical assays against a panel of 16 kinase domain mutants representing textgreater90% of clinical isolates. We report that AMN107 and BMS-354825 are 20-fold and 325-fold more potent than imatinib against cells expressing wild-type Bcr-Abl and that similar improvements are maintained for all imatinib-resistant mutants tested, with the exception of T315I. Thus, both inhibitors hold promise for treating imatinib-refractory CML.
Cancer 2005
AMN107, a novel aminopyrimidine inhibitor of p190 Bcr-Abl activation and of in vitro proliferation of Philadelphia-positive acute lymphoblastic leukemia cells.
Abstract
Abstract
BACKGROUND: Previous studies have shown that patients with Bcr-Abl-positive acute lymphoblastic leukemia (ALL) either have primary disease that is refractory to imatinib mesylate or develop disease recurrence after an initial response. METHODS: The authors investigated the effects of a newly designed Bcr-Abl inhibitor, AMN107, by comparing its in vitro inhibitory potency on p190 Bcr-Abl ALL cell lines with that of imatinib. RESULTS: In two Philadelphia (Ph)-positive ALL cell lines, AMN107 was found to be 30-40 times more potent than imatinib in inhibiting cellular proliferation. AMN107 was also more effective than imatinib in inhibiting phosphorylation of p190 Bcr-Abl tyrosine kinase in cell lines and primary ALL cells. The inhibition of cellular proliferation was associated with the induction of apoptosis in only one of the cell lines. No activity was observed in cell lines lacking the BCR-ABL genotype. CONCLUSIONS: The results of the current study suggest the superior potency of AMN107 compared with imatinib in Ph-positive ALL and support clinical trials of AMN107 in patients with Ph-positive ALL.