
Mifepristone
Progesterone and glucocorticoid receptor antagonist


概要
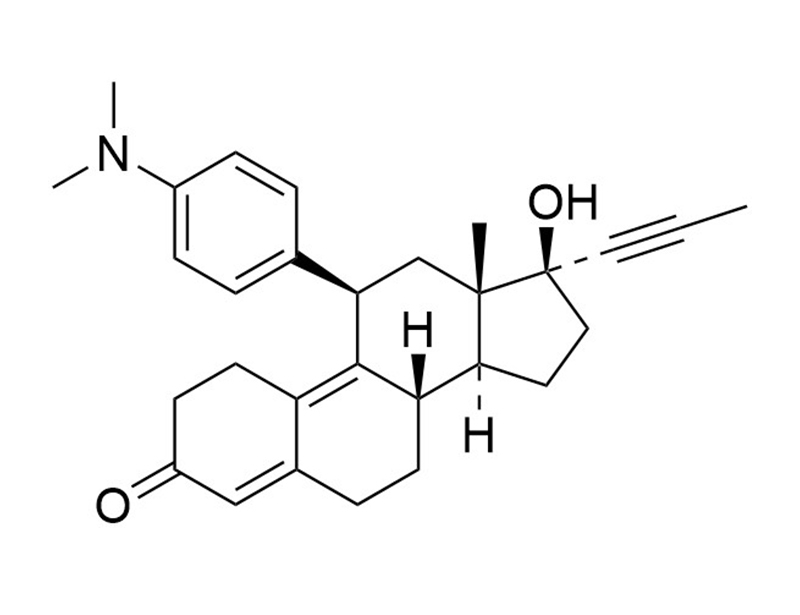
Mifepristone is a progesterone and glucocorticoid receptor antagonist (Ki = 0.64 and 0.1 nM, respectively; Attardi et al.; Song et al.; von Geldern et al.). It has been reported that Mifepristone is used in the treatment of leiomyomata, endometriosis, breast cancer, and meningioma (Mahajan et al.).
CANCER RESEARCH
· Inhibits the expressions of cyclin-dependent kinase 1 (CDK1) and cyclin-dependent kinase 2 (CDK2), leading to cell cycle arrest and apoptosis in endometrial epithelial cells and stromal cells in adenomyosis (Che et al.).
· Induces cell cycle arrest and apoptosis in antiestrogen-resistant breast cancer cells in vitro (Gaddy et al.).
CANCER RESEARCH
· Inhibits the expressions of cyclin-dependent kinase 1 (CDK1) and cyclin-dependent kinase 2 (CDK2), leading to cell cycle arrest and apoptosis in endometrial epithelial cells and stromal cells in adenomyosis (Che et al.).
· Induces cell cycle arrest and apoptosis in antiestrogen-resistant breast cancer cells in vitro (Gaddy et al.).
Alternative Names
RU-486
Cell Type
Cancer Cells and Cell Lines
Area of Interest
Cancer Research
CAS Number
84371-65-3
Chemical Formula
C29H35NO2
Molecular Weight
429.6 g/mol
Purity
≥ 98%
Target
CDK1, CDK2, Glucocorticoid Receptor
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | Mifepristone | 100-0564, 100-0565 | All | English |
| Safety Data Sheet | Mifepristone | 100-0564, 100-0565 | All | English |
数据及文献
Publications (5)
Journal of cellular and molecular medicine 2020 jan
A new trick for an old dog: The application of mifepristone in the treatment of adenomyosis.
Abstract
Abstract
Adenomyosis is also called internal endometriosis and affects about 20{\%} of reproductive-aged women. It seriously reduces life quality of patients because current drug therapies face with numerous challenges. Long-term clinical application of mifepristone exhibits wonderful therapeutic effects with mild side-effects in many disorders since 1982. Since adenomyosis is a refractory disease, we investigate whether mifepristone can be applied in the treatment of adenomyosis. In this study, we investigated the direct effects of mifepristone on human primary eutopic endometrial epithelial cells and stromal cells in adenomyosis. We found that mifepristone causes cell cycle arrest through inhibiting CDK1 and CDK2 expressions and induces cell apoptosis via the mitochondria-dependent signalling pathway in endometrial epithelial cells and stromal cells of adenomyosis. Furthermore, mifepristone inhibits the migration of endometrial epithelial cells and stromal cells through decreasing CXCR4 expression and restricts the invasion of endometrial epithelial cells via suppression of epithelial-mesenchymal transition in adenomyosis. We also found that mifepristone treatment decreases the uterine volume, CA125 concentration and increases the haemoglobin concentration in serum for adenomyosis patients. Therefore, we demonstrate that mifepristone could serve as a novel therapeutic drug in the treatment of adenomyosis, and therefore, the old dog can do a new trick.
The Journal of clinical investigation 2013 jul
Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus.
Abstract
Abstract
Recent evidence suggests that enhanced neutrophil extracellular trap (NET) formation activates plasmacytoid dendritic cells and serves as a source of autoantigens in SLE. We propose that aberrant NET formation is also linked to organ damage and to the premature vascular disease characteristic of human SLE. Here, we demonstrate enhanced NET formation in the New Zealand mixed 2328 (NZM) model of murine lupus. NZM mice also developed autoantibodies to NETs as well as the ortholog of human cathelicidin/LL37 (CRAMP), a molecule externalized in the NETs. NZM mice were treated with Cl-amidine, an inhibitor of peptidylarginine deiminases (PAD), to block NET formation and were evaluated for lupus-like disease activity, endothelial function, and prothrombotic phenotype. Cl-amidine treatment inhibited NZM NET formation in vivo and significantly altered circulating autoantibody profiles and complement levels while reducing glomerular IgG deposition. Further, Cl-amidine increased the differentiation capacity of bone marrow endothelial progenitor cells, improved endothelium-dependent vasorelaxation, and markedly delayed time to arterial thrombosis induced by photochemical injury. Overall, these findings suggest that PAD inhibition can modulate phenotypes crucial for lupus pathogenesis and disease activity and may represent an important strategy for mitigating cardiovascular risk in lupus patients.
Molecular endocrinology (Baltimore, Md.) 2004 jan
Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor.
Abstract
Abstract
Mifepristone is a potent antagonist of steroid hormone receptors such as glucocorticoid and progesterone receptors. We investigated the potential for mifepristone to act as an antiandrogen and compared it with partial androgen receptor (AR) agonists and antagonists, in particular bicalutamide. Mifepristone was an effective antiandrogen in vitro that inhibited transcription from three androgen-responsive promoters and blocked the agonist R1881 in a dose-dependent manner. Like bicalutamide, mifepristone also antagonized the action of androgen receptor with a (T877A) mutation. Mifepristone competed effectively with R1881 with a relative binding affinity comparable to that of cyproterone acetate, and much higher than that of hydroxyflutamide and bicalutamide in a binding assay. Mifepristone could effectively induce the binding of the herpes simplex viral protein 16/AR fusion protein to the hormone response elements in the murine mammary tumor virus-luciferase reporter. With either wild-type or T877A mutant AR, mifepristone alone was unable to induce any detectable interaction with coactivators transcriptional intermediary factor-2 or beta-catenin but could inhibit the R1881-induced binding of AR to transcriptional intermediary factor-2 and beta-catenin. Similarly, mifepristone could inhibit the R1881-induced N/C-terminal interaction in a dose-dependent manner even though mifepristone alone has no effect on the N/C-terminal interaction of AR. We found that mifepristone could induce a strong interaction between AR and corepressors nuclear receptor corepressor and silencing mediator for retinoid and thyroid hormone receptors in both transactivation and two-hybrid assays to a greater degree than hydroxyflutamide, cyproterone acetate, and bicalutamide. The AR-corepressor interaction was also seen in coimmunoprecipitation assays. Finally, mifepristone at high concentrations induced a low level of prostate-specific antigen expression in LNCaP and antagonized prostate-specific antigen expression induced by R1881. Mifepristone also antagonized R1881 action on the growth of LNCaP prostate cancer cells.
Journal of medicinal chemistry 2004 aug
Liver-selective glucocorticoid antagonists: a novel treatment for type 2 diabetes.
Abstract
Abstract
Hepatic blockade of glucocorticoid receptors (GR) suppresses glucose production and thus decreases circulating glucose levels, but systemic glucocorticoid antagonism can produce adrenal insufficiency and other undesirable side effects. These hepatic and systemic responses might be dissected, leading to liver-selective pharmacology, when a GR antagonist is linked to a bile acid in an appropriate manner. Bile acid conjugation can be accomplished with a minimal loss of binding affinity for GR. The resultant conjugates remain potent in cell-based functional assays. A novel in vivo assay has been developed to simultaneously evaluate both hepatic and systemic GR blockade; this assay has been used to optimize the nature and site of the linker functionality, as well as the choice of the GR antagonist and the bile acid. This optimization led to the identification of A-348441, which reduces glucose levels and improves lipid profiles in an animal model of diabetes.
Fertility and sterility 1997 dec
Mifepristone (RU486): a review.
Abstract
Abstract
OBJECTIVE To review the literature concerning the mechanism of action and pharmacodynamics of mifepristone (RU486), potential new uses of RU486, and its current use not only as an abortifacient but also as therapy for endometriosis, leiomyoma, breast cancer, and meningioma. DATA IDENTIFICATION AND SELECTION Studies that relate to RU486 were identified through a MEDLINE search. CONCLUSION(S) RU486 is an 11 beta-dimethyl-amino-phenyl derivative of norethindrone with a high affinity for P and glucocorticoid receptors. The receptor binding is not followed by transcription of P-dependent genes. Mifepristone effectively blocks P receptors in the placenta, resulting in the termination of pregnancy. In addition, it has been used in the treatment of leiomyomata, endometriosis, advanced breast cancer, and meningioma. It is a powerful tool to study the molecular action of P and in the future may be used as an estrogen-free contraceptive. Through an online search of MEDLINE, the authors reviewed the literature on the development of mifepristone (RU-486); RU-486's mechanism of action, pharmacodynamics, and distribution; the physiologic action of RU-486; potential new uses for RU-486; and its current use as both an abortifacient and therapy for endometriosis, leiomyoma, breast cancer, and meningioma. RU-486 is an 11beta-dimethyl-amino-phenyl derivative of norethindrone with a high affinity for P and glucocorticoid receptors. Receptor binding is not followed by the transcription of P-dependent genes. RU-486 effectively blocks P receptors in the placenta, resulting in the termination of pregnancy. It has also been used to treat leiomyomata, endometriosis, advanced breast cancer, and meningioma. The following therapeutic uses of RU-486 are discussed: the termination of early pregnancy, treatment with RU-486 in combination with prostaglandins, the termination of second-trimester pregnancy, cervical ripening, labor induction, postcoital contraception, uterine leiomyomata, endometriosis, breast cancer, and meningioma.